ParseCNV Copy Number
Variant Call Association Software
New GitHub Page
(ParseCNV2)
https://github.com/CAG-CNV/ParseCNV2
Linux/Mac/Windows with
Cygwin (command line)
Windows Executable
(graphical user interface)
Decompress command: gunzip ParseCNV21.tgz;
tar -xvf ParseCNV21.tar
Run Command: perl ParseCNV.pl Cases.rawcnv
Controls.rawcnv Cases.fam ChrSnp0Pos.map
On Windows, unzip, and launch by
double-clicking ParseCNV.exe (with camel icon).
Linux
bash environment is best, but on Windows Cygwin will also work.
Having
trouble? Bugs? Email Joseph Glessner: ![]()
Or post
on Google Group: https://groups.google.com/forum/#!forum/parsecnv
Input Files:
ParseCNV takes CNV calls as input and creates probe
based statistics for CNV occurrence in (cases and controls, families, or
population with quantitative trait) then calls CNVRs based on neighboring SNPs
of similar significance. CNV calls may be from aCGH, SNP array, Exome
Sequencing, or Whole Genome Sequencing.
A brief introduction: DNA base call GWAS deals with 3
possible states at each SNP [AA,AB,BB] where A and B=[A,T,C,G] and A≠B.
CNV association deals with 5 possible states at each
SNP [0,1,2,3,4] where 2 is typically high frequency as the normal diploid one
maternal one paternal copy expectation. To leverage 3 state association, we
divide out deletion [0,1,2] and duplication [2,3,4] association.
Pre and Post association quality control metrics are important
to limit bias in overly stringent Pre association quality control.
CNVs are also called Copy Number Abberations,
Changes, Differences or Polymorphisms (CNA, CNC, CND or CNP) depending on the
discipline, context and the level of specificity. CNVs are the popularly
assessed subset of Structural Variations, which include insertions and
inversions.
See simple example input files: Cases.rawcnv Controls.rawcnv
Cases.fam ChrSnp0Pos.map IDToPath.txt and try to run them as a test of the
software on your system. If you have trouble with your input files, create
similarly simple versions to quickly run the software and isolate the problem.
Run Command: perl
ParseCNV.pl Cases.rawcnv Controls.rawcnv Cases.fam ChrSnp0Pos.map (optional
--out <prefix>, --build <hg18>, --idToPath <file.txt>,
--includePed, --probesBafLrr <file.txt>, --batch <file.txt>,
--mergePVar <1>, --mergeDist <1000000>, --tdt, --maxPInclusion
<0.05>, --includeAllRedFlags)
Dependencies: bash, R,
and plink. Try which bash, which R, and which plink and if any returns command
not found instead of path, try: "module avail" and "module load
R" otherwise download and install these dependencies then do "export
PATH=/YourDirectoryPath/R-3.1.2/bin:$PATH".
Read and cite the paper:
New Major Features
(full version log at bottom):
NEW Feature! 2015-05-14 --includePed now writes
directly to a Plink .bed file to save disk space
NEW Feature! 2014-11-03 ParseCNV_GUI_Windows:
Executable Minimized Memory for big data but a bit slower
NEW Feature! 2014-02-27 MakeRedFlagPlots.pl: derive
continuous confidence score based on redFlagReasons column in ParseCNV
--includeAllRedFlags
NEW Feature! 2014-02-22 PQN_Concord_Inh.pl: multiple
CNV detection algorithm comparison and de
novo, transmitted, and untransmitted CNV call based annotation
NEW Feature! 2013-12-13 Graphical Interface for
ParseCNV.pl using wxPerl and Tk ParseCNV_GUI.pl
NEW Feature! 2013-11-25 --build bosTau7 (Cow 1-29 X)
--build susScr3 (Pig 1-18 XY) now supported in addition to hg18 hg19
NEW Feature! 2013-11-21 Brief column .stats main
folder output, ParseCNV_QC.pl QC Thresholds may be specified, basic non-human
species support
NEW Feature! 2012-11-27 Sample quality control
metrics and sample remove report now automated in ParseCNV_QC.pl
NEW Feature! 2012-11-25 Gene based association
approach now available in ParseCNV_GeneBased.pl
NEW Feature! 2012-11-20 BAF/LRR plots of two samples
with associated CNV and one sample with CN=2 with flanking diploid data for
each CNVR with --idToPath
NEW Feature! 2012-08-25 Image created with BAF and
LRR of specific association regions across contributing cases as part of
--idToPath option.
NEW Feature! 2012-04-09 Plink p values can now be
imported into ParseCNV using InsertPlinkPvalues.pl for CNVR boundary calling.
NEW Feature! 2012-02-06 UCSC review features are now
automatically scored.


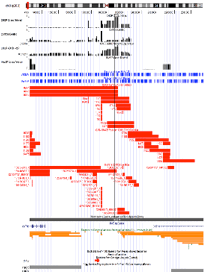
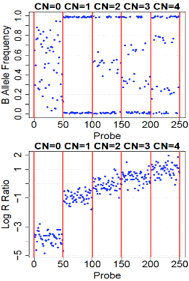
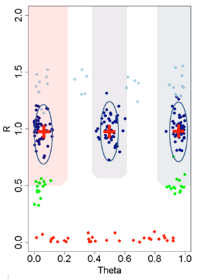
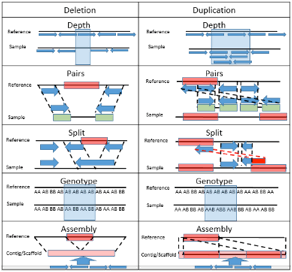
CNV Calls
CNV calls (.rawcnv) can
be generated by any algorithm but should be in the PennCNV format:
CNVCallChromosome:Start-StopBoundaries NumProbes
Size(BasePairs) CNState SampleID StartSNP EndSNP
Format conversion scripts for popular algorithms (QuantiSNP, XHMM, and GenomeStrip) are included. Typically this reformatting is relatively easy in Excel. Calls for a given sample should be sorted together in subsequent rows. CNState values: state1,cn=0 state2,cn=1 state5,cn=3 state6,cn=4. These states are also referred to as homozygous deletion, hemizygous deletion, hemizygous duplication, and homozygous duplication respectively in literature. Be careful there is no blank line at end of file. PennCNV states [1,2,5,6] correspond to CN [0,1,3,4].
For example:
“chr1:156783977-156788016 numsnp=6
length=4,040
state2,cn=1 sample1.txt startsnp=rs16840314 endsnp=rs10489835”
Alternatively: “chr1:156783977-156788016 numsnp=6
length=4,040
state2,cn=1 sample1.txt startsnp=rs16840314 endsnp=rs10489835 conf=20.659”
There are separate files for the study subjects
(cases and related parents or siblings if available) and control subjects.
If there are no
control subjects as in a family based or quantitative trait association study,
use the file NullControl_ForQuantitativeTrait.rawcnv for the Controls.rawcnv
input.
If you want to filter a .rawcnv with both cases and
controls into separate input files you can use: GeneRef/RemoveSpecificBarcodes-Hash_Example_DefFile_ColRemKeep.pl
(FilterCNV.pl)
state3,cn=2 on chrX is male duplication so should be
recoded state5,cn=3
state4,cn=2 means copy neutral LOH i.e. ROH
BED (4 columns: chr start stop id) TO RAWCNV: awk
'{print "chr"$1":"$2"-"$3"\tnumsnp=1\tlength="$3-$2"\tstate2,cn=1\t"$4"\tstartsnp="$1"_"$2"\tendsnp="$1"_"$3}'
human_g1k_v37.mask.36.bed > human_g1k_v37.mask.36.rawcnv
RAWCNV TO Plink CNV: sed 's/:/\t/' Cases_wConf.rawcnv
| sed 's/-/\t/' | sed 's/chr//' | sed 's/state.\,cn\=//'| sed 's/conf=//' | sed
's/numsnp=//' | awk '{print "0 "$7" "$1"
"$2" "$3" "$6" "$10" "$4}'
RAWCNV TO VCF:
sed 's/:/\t/' 201904530099_R08C01.rawcnv | sed 's/-/\t/' | awk
'{ORS=""; if(NR==1)print
"##fileformat=VCFv4.1\n#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t"$7"\n";
print
$1"\t"$2"\t"$1":"$2"-"$3"\t<DIP>\t";
if($6~"cn=0"||$6~"cn=1"){print
"<DEL>"}else{print "<DUP>"}print
"\t.\t.\tEND="$3";"$4";"$5";"$6";"$8";"$9";"$10"\tGT\t";
if($6~"cn=0"||$6~"cn=4"){print "1/1\n"}else{print
"0/1\n"}}' > 201904530099_R08C01.vcf
PennCNV: compile_pfb.pl creates a .pfb and cal_gc_snp.pl
creates a .gcmodel for your specific array. Use the same version of array for
cases and controls.
Please make sure you use --log (or
2>>MySamples.log) and --conf command line arguments for quality
assessment. Then:
grep summary MySamples.log > MySamples_QC.txt
awk ‘{print $5}’ MySamples.rawcnv | uniq --count > MySamples_CountCNVs.txt
If you want ChrX and LOH(copy neutral LOH/ROH) calls,
you have to run separately --chrx and --loh --hmm hh550.hmm
Merging fragmented calls with clean_cnv.pl is useful,
especially if you are interested in large CNVs: perl clean_cnv.pl combineseg file.rawcnv
-signalfile file.pfb -out file_clean.rawcnv
PFB gives universal definition for snp to chr pos and
snp set used
detect_cnv.pl --trio after detect_cnv.pl --test is
strongly recommended if you have trios to limit the de novo rate.
Review LRR BAF of the parents for false negative CNVs
by copying child CNVs and adding parent IDs and state3,cn=2 to run
visualize_cnv.pl the run: convert Mom.JPG Dad.JPG +append Parents.JPG; convert
Parents.JPG Child.JPG -append Trio.JPG
|
.signalfile (.baflrr) .pfb
.hmm .gcmodel |
PennCNV ---------------> |
.rawcnv .log .fam .map |
ParseCNV ---------------> |
.cnvr .stats |
PennCNV-ExomeSeq: PennCNV Copy Number Variant Call
Detection in Exome Sequencing Software http://penncnvexomeseq.sourceforge.net
QuantiSNP: You have to download from here. The various other websites do not work. Make sure your files are: “SNPName Chromosome
Position LRR BAF” the header is not read and if you switch LRR and BAF no error
is given.
Plink: Run --missing to get call rates
(or from GenomeStudio/GenotypingConsole), --genome to check relatedness (high
PI_HAT), --mendel to verify families, --check-sex to verify gender, .mds (--genome
then --read-genome --cluster --mds-plot) or Eigenstrat smartpca to create
.pca.evec for ethnicity/stratification.
GenomeSTRiP: Always use the reference metadata bundles and
download using wget NOT drag and drop (which gives a truncated file):
wget ftp://ftp.broadinstitute.org/pub/svtoolkit/reference_metadata_bundles/1000G_phase1_12May2015.tar.gz
wget ftp://ftp.broadinstitute.org/pub/svtoolkit/reference_metadata_bundles/1000G_phase3_12May2015.tar.gz
|
.fastq .fasta |
BWA-MEM --------------> -M |
.bam Ref Align CN2 Ploidy Gender |
GenomeSTRiP ------------------> (SVPreprocess, SVDiscovery, SVGenotyper,
CNVDiscoveryPipeline NOT SVAltAlign) |
test1.genotypes.vcf cnv_stage12/gs_cnv.genotypes.vcf.gz |
ConvertVCFToPennCNV_GenomeStrip.pl ---------------------------------------------------------->
|
.rawcnv |
ParseCNV -------------> |
.cnvr |
If you only have
PED/BED genotype file, you need to use the Illumina Genome Studio Project with the Idats from
your samples loaded and export BafLrr files. Use column chooser to show B
allele Freq and Log R Ratio along with SNP Name, Chr, and Position. Use the
PennCNV packaged script kcolumn.pl to convert the all samples text file into
single sample text files. See detailed instructions here:
http://penncnv.openbioinformatics.org/en/latest/user-guide/input/ If you are
using Affymetrix, see here:
http://penncnv.openbioinformatics.org/en/latest/user-guide/affy/ Once you have
generated a PennCNV .rawcnv file (using the --conf option), you can convert
into a Plink CNV file using a simple bash command: sed 's/:/\t/'
Cases_wConf.rawcnv | sed 's/-/\t/' | sed 's/chr//' | sed 's/state.\,cn\=//'|
sed 's/conf=//' | sed 's/numsnp=//' | awk '{print "0 "$7"
"$1" "$2" "$3" "$6" "$10"
"$4}' or use ParseCNV which uses PennCNV .rawcnv as input directly. See
Plink CNV format specification here:
http://pngu.mgh.harvard.edu/~purcell/plink/cnv.shtml
Otherwise, it is very hard to make any meaningful
inference of CNVs from a SNP genotype PED file. We can infer CNV by intensity
(LRR) alone, but not by genotype alone.
The best you could do is detect runs of homozygosity
using plink:
plink --ped file.ped --map file.map --homozyg
http://pngu.mgh.harvard.edu/~purcell/plink/ibdibs.shtml#homo
This would flag regions, which could be deletions
(with intensity drop) or simply runs of homozygosity (which do not have the
intensity drop). You can convert the plink ROH output into a plink CNV input
file using Excel or awk. You can just put Type as 1 to presume they are mostly
single copy deletions.
You could similarly look for "runs of
no-calls" which would flag regions of homozygous deletion or duplication,
again non-differentiatable without the intensity data.
This is the ambiguity you would have to accept given
the limited data available.
Some papers say you can impute CNVs from SNP data,
but I am skeptical and at best would only detect mostly common CNVs, which are
typically of less interest.
FAM
The family or FAM file has the family relationships
in the study subjects to track inheritance of CNVs. Tab Delimited:
FamID
IndividualID(InCNVCallFile) FatherID MotherID Gender(1=male,2=female) Affected(1=unaffected,2=affected,-9=exclude).
IndividualID and Affected columns are required others
can be zeroed if not applicable.
For example: “1 1.txt 0 0 1 2”
If you have Cases.rawcnv and they are just unrelated
cases, without any families (unrelated), or quantitative traits you can just
run: awk '{print $5}' Cases.rawcnv | uniq | sed 's/^/0\t/' | sed
's/$/\t0\t0\t0\t2/' > Cases.fam
MAP
The SNP definition file is four columns:
Chromosome ProbeID
PositionCentiMorgan PositionBasePair
similar to Plink map file (PositionCentiMorgan is
fine just zeroed out). Sorted by position and chromosome Sorted.map created automatically
which should be used with the resulting ped. Map could be an intersection or
union of chip types if study is across platforms. Be sure that all SNPs used
for CNV calling are provided.
For example: “1 rs1739822 0 16196820”
If you are using sequencing
data or are uncertain about the full set of probes, a blank or incomplete file
can be used as the map input and the map will be dynamically determined by the
start and end positions (breakpoints) of the CNVs observed in the .rawcnv
files.
The full .map chr snp pos information should be
available on the Illumina/Affymetrix website, baflrr "signal intensity
files" PennCNV inputs, in Illumina GenomeStudio / Affymetrix Genotyping Console,
or PennCNV .pfb file. You can use the linux awk command to rearrange the
columns if needed.
Chr X should be X, not 23 as in some plink processed
files.
For example if you have the PennCNV .pfb run: awk
'{print $2"\t"$1"\t0\t"$3}' ChipX.pfb > ChipX.map
If you have a .vcf run: head -1
DATA.PCA_normalized.filtered.sample_zscores.RD.txt | sed 's/\s/\n/g' | grep -v
Matrix | sed 's/:/\t/' | awk '{print
$1"\t"$1"_"$2"\t0\t"$2}' > file.map
OR awk '{print $1" "$2}' file.vcf | sed
's/^chr//' | awk '{print $1"\t"$1"_"$2"\t0\t"$2}'
| grep -v ^# > file.map
ParseCNV will automatically define probes missing in
the map according to the .rawcnv definitions but this should only be utilized
for a small number of probes to properly represent the actual data resolution.
CHR_POS TO MAP: awk '{print
$1"\t"$1"\t0\t"$1}' b | sed 's/_/\t/g' | awk '{print
$1"\t"$3"_"$4"\t0\t"$7}' | sort -n -k 4,4 | sort -k
1,1 -s > b_Sorted.map
Download Test Data 785 cases, 1110 controls,
561,308 probes
Out of Memory:
Try running per chr by
“grep chr1: file.rawcnv” if runs out of memory indicated by percentage updating
getting cutoff.
grep chr1: Cases.rawcnv > Cases.rawcnv_chr1
grep chr1: Controls.rawcnv > Controls.rawcnv_chr1
grep -P "^1\s" ChrSnp0Pos.map > ChrSnp0Pos.map_chr1 OR sed 's/^/chr/'
ChrSnp0Pos.map | awk '{print
$1":\t"$2"\t"$3"\t"$4}' | grep chr1: | sed
's/chr//' | sed 's/://' > ChrSnp0Pos.map_chr1
Cases.fam no edit needed
cat SSC_ParentsControlOnly_PCGC_chr1_Recluster_CNVR_ALL_ReviewedCNVRs.txt
outputs when done
--splitByChr --cluster --sub bsub --clusterHeader
MGH_Header.txt
--splitByRange --cluster --sub bsub --clusterHeader
MGH_Header.txt
ParseCNV_BigData_LowMemory_Slower.pl
--out output
file prefix.
--idToPath <file.txt> Additional
SampleID to BafLrrFilePath file for Specific BAF LRR access of all associated
samples for associated CNVRs. The input file should contain 2 columns: IDs as
in column 5 of .rawcnv and full path to baf lrr file (signal intensity). In
many cases, the 2 columns may be the same.
--probesBafLrr <file.txt> If Probe ID not first
column of BAF LRR file, provide probe order reference file.
--includePed write .ped file for usage in PLINK
--build <hg18> Specify
the genome build used in CNV calling and .map file. Uses corresponding build
files in GeneRef folder. See README.txt end for build specific download
commands (example below).
--batch <file.txt> A
subset of samples which form a discernible batch to monitor the contributions
of such as a different chip type/version, processing lab, sample type/source,
or reagent batch.
--mergePVar <1> Change
the default of merging probes with p-values no more than one power of ten
variation.
--mergeDist <1000000> Change the default of merging
probes with proximal distance no more than 1000000 base pairs (1MB). The
default is good for 500k but for 2.5M since 5x more markers, use 1/5 of 1M MB =
200,000.
--tdt Specify
a family based study with trios to allow TDT deletion and duplication p-value
to drive CNVR boundary determination rather than case:control.
--maxPInclusion <0.05> The maximum p value of probe based statistics to
include in CNVR determination and thus reported associations.
--includeAllRedFlags Include
all red flag values in RedFlagReasons regardless of extreme values
--keepTemp Keep
temp folder files (mainly for Verbose.stats currently dependency of
InsertPlinkPValues.pl)
--splitByChr Split
run by chromosome for large projects that run out of memory
--splitByRange Split
run by equal sized ranges for large projects that run out of memory
--cluster option
requires a parallel computing cluster you can qsub to (speeds up split by Chr
run)
--lowRange <0> low
index for range specific run if only interested in specific locus or for large
memory intensive projects
--highRange <nProbes> high index for range specific
run if only interested in specific locus or for large memory intensive projects
--oneSidedP calculate
one sided P assuming only enriched in case CNVs are of interest (default two
sided)
--permuteP <1000> calculate
permuted P providing the number of permutations (takes longer time
computationally)
|
Explanation |
Category |
|
|
ParseCNV_GeneBased.pl |
Gene based association approach counting any overlap of
gene or gene exons together (collapsing association strategy) |
Collapsing Approach Association Alternative to
ParseCNV.pl |
|
Polishgc5BaseForScanRegion_KeepOnlyChrLines_5120Increments.pl |
Make GC file format for scan_region.pl for non-human
species in 5120 increments to be similar to the human definition |
definition utility |
|
PolishRefGeneForScanRegion_KeepOnlyChrLines.pl |
Make RefGene file format for scan_region.pl for
non-human species keeping only "Chr Number" entries |
definition utility |
|
AvgGCFromScanRegion.pl |
Averages GC |
dependency of AnnotateCNV |
|
CountAvgScanRegion.pl |
Counts DGV and SegDups and Averages GC as Part of
AnnotateCNV.txt Commands |
dependency of AnnotateCNV |
|
1_A.baflrr |
Example BafLrr SignalFile |
example file |
|
2_A.baflrr |
Example BafLrr SignalFile |
example file |
|
3_A.baflrr |
Example BafLrr SignalFile |
example file |
|
4_A.baflrr |
Example BafLrr SignalFile |
example file |
|
5_A.baflrr |
Example BafLrr SignalFile |
example file |
|
Batch.txt |
Example Batch input file |
example file |
|
Cases.fam |
Example Fam File |
example file |
|
Cases.rawcnv |
Example Cases rawcnv file |
example file |
|
ChrSnp0Pos.map |
Example Map File |
example file |
|
Controls.rawcnv |
Example Controls rawcnv file |
example file |
|
IDToPath.txt |
Example rawcnv column 5 ID matched to BafLrr SignalFile
path (in many cases will be the same) |
example file |
|
NullControl_ForQuantitativeTrait.rawcnv |
Empty Controls rawcnv file if running quantitative trait
where all subjects are in Cases.rawcnv |
example file |
|
probesBafLrr.txt |
Example probesBafLrr input file in case BafLrr
SignalFiles do not have SNP ID column but are just in a standard order |
example file |
|
WeightTEST.pheno |
Example Quantitative Trait Phenotype File |
example file |
|
GeneRef |
Genome definition files from UCSC Tables |
folder definition files |
|
ArrayConcordance_InheritanceDenovo_AnnotateCNVCalls |
PQN_Concord_Inh.pl: multiple CNV detection algorithm
comparison and de novo, transmitted, and untransmitted CNV call based
annotation |
folder denovo multiple algorithms |
|
PerlModules |
Perl Modules and scripts used within the main folder
scripts |
folder perl modules |
|
ParseCNV_GUI_Tk.pl |
Graphical user interface for ParseCNV.pl using perl Tk |
GUI ParseCNV.pl |
|
ParseCNV_GUI_Wx.pl |
Graphical user interface for ParseCNV.pl using perl Wx |
GUI ParseCNV.pl |
|
ConvertQuantiSNPToPennCNV.pl |
Convert QuantiSNP format CNV Calls to PennCNV format |
input conversion |
|
ConvertVCFToPennCNV_GenomeStrip.pl |
Convert GenomeStrip VCF format CNV Calls to PennCNV
format |
input conversion |
|
ConvertVCFToPennCNV_XHMM.pl |
Convert XHMM VCF format CNV Calls to PennCNV format |
input conversion |
|
ParseCNV_QC.pl |
Sample Quality Control metrics plotting and sample
remove report based on automatic or manual thresholds |
input utility |
|
TestConcordance.pl |
compares pairs of CNV calls |
input utility |
|
FilterCNV.pl |
Handy utility to keep or remove a certain list of
samples from a specified column, such as QC filtering the .rawcnv file |
input utility |
|
AnnotateCNV.txt |
Annotates many of the genomic feature red flags onto
.rawcnv calls |
output utility |
|
CountGenes.pl |
Counts the number of values in a comma separated list
such as genes |
output utility |
|
FindSnpsInRange.pl |
Query a genomic region of interest in the ParseCNV
.stats file |
output utility |
|
GetSnpsInSnpsRange.pl |
Query Multiple genomic regions of interest in the
ParseCNV .stats file |
output utility |
|
InsertPlinkPvalues.pl |
Use Plink or other software p-values to define ParseCNV
CNVRs and RedFlag Annotations |
output utility |
|
MakeRedFlagPlots.pl |
Derive continuous confidence score based on
redFlagReasons column in ParseCNV --includeAllRedFlags |
output utility |
|
PercentSamples.pl |
Showing contributing
counts of batches in IDsCasesDel/IDsCasesDup column. |
output utility |
|
wgetUcscPdfs.sh |
Pulls UCSC Track high-quality images for review of
significant CNVRs |
output utility |
|
ParseCNV.pl |
THE MAIN SCRIPT. |
ParseCNV.pl |
|
extremevalues_2.2.tar.gz |
Used by ParseCNV_QC R module to find extreme values
(needs work, perhaps substitute median absolute deviation) |
perl module (could move into folder) |
|
ParseCNV_Denovo.pl |
Reformat files for de novo calling (probably not needed) |
probably not needed |
|
PennCNV_MultSpaceToTab.pl |
Used by ParseCNV_Denovo (probably not needed) |
probably not needed |
|
README.txt |
Standard Readme file, most pertinent updated information
will be on the website |
README |
Download hg19/hg38 definition files
by pasting these commands (Included in standard download now):
cd GeneRef
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/knownGene.txt.gz
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/kgXref.txt.gz
wget
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/bioCycPathway.txt.gz
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/keggPathway.txt.gz
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/keggMapDesc.txt.gz
wget
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/cgapBiocPathway.txt.gz
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/cgapAlias.txt.gz
wget
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/cgapBiocDesc.txt.gz
gunzip knownGene.txt.gz; mv knownGene.txt hg19_knownGene.txt
gunzip kgXref.txt.gz; mv kgXref.txt hg19_kgXref.txt
gunzip bioCycPathway.txt.gz; mv bioCycPathway.txt hg19_bioCycPathway.txt
gunzip keggPathway.txt.gz; mv keggPathway.txt hg19_keggPathway.txt
gunzip keggMapDesc.txt.gz; mv keggMapDesc.txt hg19_keggMapDesc.txt
gunzip cgapBiocPathway.txt.gz; mv cgapBiocPathway.txt hg19_cgapBiocPathway.txt
gunzip cgapAlias.txt.gz; mv cgapAlias.txt hg19_cgapAlias.txt
gunzip cgapBiocDesc.txt.gz; mv cgapBiocDesc.txt hg19_cgapBiocDesc.txt
perl MakeExonScanRegionDefFile.pl hg19_knownGene.txt
perl MakeExonScanRegionDefFile_wGene.pl hg19_knownGene.txt
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/cytoBand.txt.gz
gunzip cytoBand.txt.gz; mv cytoBand.txt hg19_cytoBand.txt
awk -F"\t" '{print
$1$4"\t"$1"\t+\t"$2"\t"$3"\t \t \t \t \t \t
\t "}' hg19_cytoBand.txt | sed 's/chr//' > hg19_cytoBand_SimFormat_AllCol.sorted
wget
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/genomicSuperDups.txt.gz
gunzip genomicSuperDups.txt.gz
sort -k 2,2 -k 3,3n genomicSuperDups.txt > hg19_genomicSuperDups.sorted
awk '{print $27"\t"$2"\t"$7"\t"$3"\t"$4"\t
\t \t \t \t \t \t "}' hg19_genomicSuperDups.sorted >
hg19_genomicSuperDups_SimFormat_AllCol.sorted
rm genomicSuperDups.txt
rm hg19_genomicSuperDups.sorted
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/dgvMerged.txt.gz
gunzip dgvMerged.txt.gz; mv dgvMerged.txt hg19_dgv.txt
awk -F"\t" '{print $12"\t"$2"\t"$7"\t"$3"\t"$4"\t
\t \t \t \t \t \t "}' hg19_dgv.txt> hg19_dgv_SimFormat_AllCol.sorted
rm hg19_dgv.txt
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/gc5Base.txt.gz ## wget http://hgdownload.cse.ucsc.edu/gbdb/hg38/bbi/gc5BaseBw/gc5Base.bw
gunzip gc5Base.txt.gz
awk -F"\t" '{print
$13/$12"\t"$2"\t+\t"$3"\t"$4"\t \t \t \t \t
\t \t "}' gc5Base.txt> hg19_gc5Base_SimFormat_AllCol.sorted
rm gc5Base.txt
hg19_somaticRearrangement_SimFormat_AllCol.sorted now
provided in download if error arises at that step.
Was: dgv.txt.gz, now:dgvMerged.txt.gz and dgvSupporting.txt.gz
Quantitative Trait Association
§
run
ParseCNV with --includePed and --out <myPrefix> (Cases.rawcnv can have
all samples and Controls.rawcnv can just be a blank file with no blank line at
end NullControl_ForQuantitativeTrait.rawcnv)
§
run
plink --ped CNVDEL.ped --map myMap_UseWithPed.map --make-bed --out CNVDEL_Bed
and again with CNVDUP.ped (this will compress and allow .fam to be easily
edited, just make sure you keep the sorted order of .fam the same)
§
run
plink --bfile CNVDEL_Bed --pheno file.pheno --assoc --adjust --all-pheno --out
CNVDEL_Bed_Qassoc --allow-no-sex and again with CNVDUP_Bed (see here and here)
§
perl
InsertPlinkPvalues.pl --del <Plink deletion association result file
.qassoc> --dup <Plink duplication association result file .qassoc>
--outPrefix <ParseCNV out Prefix from first run with --includePed>
§
Note
many SNPs do not have any CNV frequency with “NA” p-values or low/rare CNV
frequency.
§
Be sure your plink input
and output files are all in the ParseCNV folder. The .pheno file should
be: FID IID QuantitativeTrait
§
InsertPlinkPvalues.pl
will then generate the CNVR reports for quantitative trait association instead
of ParseCNV.pl which generates CNVR reports for Case-Control and TDT.
§
Example:
§
Cases.rawcnv
in this case contains all samples in your study since all subjects have a
continuous trait like weight and are not easily separated into simple case and
control cohorts.
§
perl
ParseCNV.pl Cases.rawcnv NullControl_ForQuantitativeTrait.rawcnv Cases.fam
ChrSnp0Pos.map --includePed --out TEST
§
plink
--ped TESTCNVDEL.ped --map TESTChrSnp0PosSorted_UseWithPed.map --pheno
WeightTEST.pheno --out DelWeightTEST --noweb --assoc
§
plink
--ped TESTCNVDUP.ped --map TESTChrSnp0PosSorted_UseWithPed.map --pheno
WeightTEST.pheno --out DupWeightTEST --noweb --assoc
§
perl
InsertPlinkPvalues.pl --del DelWeightTEST.qassoc --dup DupWeightTEST.qassoc
--outPrefix TEST --out TESTInsert
§
To
correct for population variation, include “--covar myMDSResult.mds” or “--covar
myPCAResult.pca.evec” to the plink command as detailed below. (Note
–assoc and --tdt do not work with --covar so use --linear or --logistic
instead.)
§
If you want to use other P-value
generating tools such as GEMMA update the header to be “SNP”, “BETA” or “T” or “OR”,
and “P”, just note InsertPlinkPvalues dependencies
§
REQUIRED DEPENDENCY:
§
TESTCNV_Verbose.stats
§
CAN MAKE FILES:
§
vim TESTParseCNV.log containing
“Running Command: perl ParseCNV.pl Cases.rawcnv Controls.rawcnv Cases.fam
ChrSnp0Pos.map --includePed --out TEST”
§
vim DelWeightTEST.log
§
vim DupWeightTEST.log
Population Stratification Correction
§
Use
genotype .ped in Plink to create .mds result (--genome then --read-genome
--cluster --mds-plot 3 or 10) or in Eigenstrat smartpca to create .pca.evec
result.
§
run
ParseCNV with –includePed (which now creates Individual-major
bed/bim/fam)
§
run
plink --bed plinkDel.bed --bim myMap_UseWithPed.bim --fam ALLFAM.fam
--make-bed --out CNVDEL and again with --bim plinkDup.bed --out CNVDUP changed
(which creates SNP-major bed/bim/fam)
§
run
plink --bfile CNVDEL --covar myMDSResult.mds --logistic --adjust --allow-no-sex
and again with CNVDUP
§
egrep
‘SNP|ADD’ MDS.assoc.logistic > MDS.assocADDlogistic (important to name this
way without extra “.” so Plink .log found, If you are using Affy “SNP_” IDs
choose another word unique to the header)
§
InsertPlinkPvalues.pl
--del <Plink deletion association result file> --dup <Plink
duplication association result file> --outPrefix <ParseCNV out Prefix from
first run with --includePed>
§ I recommend checking for CaseControl
PCA overlap -> Majority Cluster (typically Caucasians) ensuring overlap
-> Logistic -> then separately do African and Asian analysis since
ethnicity false positives may occur for rare CNVs and direction of effect may
differ
§ I support one quantitative trait
specified in the .pheno. I do not currently support QuantitativeTrait in the
.fam affected column or multiple quantitative traits in the .pheno.
§ You can also use LMM,
which is agrued to be the best stratification correction method for rare variants
such as CNVs
§ Gemma has this command
to generate the relatedness/kinship matrix: ./gemma -bfile
/work/1a/SCZ/chr1CNVDEL -gk 1 -o chr1CNVDELGemmaKinship
§ Then you run Gemma again
to do the actual p-value calculations: ./gemma
-bfile /work/1a/SCZ/chr1CNVDEL -k ./output/chr1CNVDELGemmaKinship.cXX.txt -lmm
4 -maf 0.000000005 -o chr1CNVDELGemma
Collapsing approach used to combine non-overlapping
CNVs which all overlap the same gene.
Gene based association approach now available in
ParseCNV_GeneBased.pl
perl
ParseCNV_GeneBased.pl Cases.rawcnv Controls.rawcnv
sort by OR>1 for Case enriched significant Genes.
The gene based approach will give many more potential
hits because it is more flexible and sensitive. Not as many features are there
but it is useable. It should work in a similar way for METAL with all necessary
input columns.
The gene-based association collapses CNVs based on
overlapping any sub-segment of the gene rather than the same sub-segment as in
the case of a CNVR defined by ParseCNV. Thus one case may overlap exon 1 of
gene x while another case may overlap exon 2 of gene x and yield significance
in a gene-based association but not in a traditional CNVR SNP-based statistics
strict overlap association.
http://www.nature.com/nature/journal/v459/n7246/extref/nature07953-s1.pdf
Supplementary Figure 5 and p37 lower panel
Burden
See Burden.txt
output from ParseCNV.pl
We need to filter the CNV calls by PennCNV provided
[numsnp, length, and conf], run PennCNV clean_cnv.pl --fraction 0.8 to merge
fragmented calls, genomic features [seg dups, DGV, Telo/Centromere/GC/somatic
rearrangements], array features[sparse coverage/low AB frequency in dups],
cohort features[population frequency]. Finally do some spot confirmation by
BAF/LRR review and PCR validation.
As always, check what chip versions make up the data
and if the full probe set was used.
The total length burden doesn't take into account the
number of CNVs or the number of genes overlapped by those CNVs.





I have tested bosTau7 Cow and susScr3 Pig
specifically to work. If you have a different species or build you can try
replacing “bosTau7” in the commands below. Mouse,
chimp, and fly should work. You may need to run the UCSC LiftOver web tool if
definition files are not available in your build version.
Segmental Duplications annotations available through
UWash Eichler website: http://eichlerlab.gs.washington.edu/database.html has: Mouse, chimp, rat, dog, fly, worm, monkey, fugu, chicken, cow, and stickleback.
Database of Genomic Variants (DGV) only has healthy human
control CNVs.
dbVar is similar for more species ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/data/ has: Bos_taurus cow, Canis_lupus gray wolf, Danio_rerio Zebrafish, Drosophila_melanogaster fly, Equus_caballus Horse,
Homo_sapiens Human, Macaca_mulatta Rhesus macaque, Mus_musculus Mouse, Pan_troglodytes chimpanzee, Sorghum_bicolor grass grain, and Sus_scrofa Pig.
Download bosTau7 Cow definition
files by pasting these commands:
cd GeneRef
wget http://hgdownload.cse.ucsc.edu/goldenPath/bosTau7/database/refGene.txt.gz
wget http://hgdownload.cse.ucsc.edu/goldenPath/bosTau7/database/refLink.txt.gz
###wget http://hgdownload.cse.ucsc.edu/goldenPath/bosTau7/database/gc5BaseBw.txt.gz
wget http://hgdownload.cse.ucsc.edu/gbdb/bosTau7/bbi/gc5Base.bw
###Use
hg19 Pathways files by querying TOUPPER of all genes
wget http://hgdownload.cse.ucsc.edu/goldenPath/bosTau7/database/cytoBandIdeo.txt.gz
wget http://cowparalogy.gs.washington.edu/data/cow4GenomicSuperDupgenomicSuperDup.tab ###NoHeader SeeHeader in
http://paralogy.gs.washington.edu/build37/data/GRCh37GenomicSuperDup.tab
wget
ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/data/Bos_taurus/by_assembly/Btau_4.6.1/gvf/Btau_4.6.1.remap.all.germline.ucsc.gvf.gz
gunzip refGene.txt.gz; mv refGene.txt
bosTau7_refGene.txt
gunzip refLink.txt.gz; mv refLink.txt
bosTau7_refLink.txt
###gunzip gc5BaseBw.txt.gz; mv gc5BaseBw.txt
bosTau7_gc5BaseBw.txt
perl PolishRefGeneForScanRegion_KeepOnlyChrLines.pl
bosTau7_refGene.txt ### to delete non “chr” lines
perl MakeExonScanRegionDefFile_refGene.pl
bosTau7_refGene.txt_OnlyChr.txt
perl MakeExonScanRegionDefFile_wGene_refGene.pl
bosTau7_refGene.txt_OnlyChr.txt
#cp PerlModules/scan_region.pl
.
wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/bigWigToBedGraph
chmod 777 bigWigToBedGraph
###
These steps will take considerable compute time
wget http://hgdownload.cse.ucsc.edu/gbdb/bosTau7/bbi/gc5Base.bw
mv gc5Base.bw
bosTau7_gc5Base.bw
./bigWigToBedGraph bosTau7_gc5Base.bw
bosTau7_gc5Base.bedGraph
perl Polishgc5BaseForScanRegion_KeepOnlyChrLines_5120Increments.pl
bosTau7_gc5Base.bedGraph
###
Illegal division by zero at
Polishgc5BaseForScanRegion_KeepOnlyChrLines_5120Increments.pl line 30,
<SAMPSHEET> line 358024431.# so no Y
awk -F"\t"
'{print $4"\t"$1"\t+\t"$2"\t"$3"\t \t \t \t
\t \t \t "}' bosTau7_gc5Base.bedGraph_OnlyChr_5120Intervals.txt >
bosTau7_gc5Base_SimFormat_AllCol.sorted ### Now unique values, rand() was added
before to $4 GC to allow duplicates in scan region since original 5 base pair
interval values are only 20,40,60,80
###perl Polishgc5BaseForScanRegion_KeepOnlyChrLines.pl
bosTau7_gc5Base_SimFormat_AllCol.sorted_randDec
rm bosTau7_gc5Base.bw; rm bosTau7_gc5Base.bedGraph; rm bosTau7_gc5Base.bedGraph_OnlyChr_5120Intervals.txt
gunzip cytoBandIdeo.txt.gz; mv cytoBandIdeo.txt bosTau7_cytoBandIdeo.txt
awk -F"\t" '{print
$1$4"\t"$1"\t+\t"$2"\t"$3"\t \t \t \t \t \t
\t "}' bosTau7_cytoBandIdeo.txt | sed 's/chr//' > bosTau7_cytoBand_SimFormat_AllCol.sorted
mv
cow4GenomicSuperDupgenomicSuperDup.tab bosTau7_cow4GenomicSuperDupgenomicSuperDup.tab
sort -k 1,1 -k 2,2n
bosTau7_cow4GenomicSuperDupgenomicSuperDup.tab >
bosTau7_cow4GenomicSuperDupgenomicSuperDup.tab.sorted #Sort By Chr Pos
awk '{print
$7”:”$8"\t"$1"\t"$6"\t"$2"\t"$3"\t
\t \t \t \t \t \t "}'
bosTau7_cow4GenomicSuperDupgenomicSuperDup.tab.sorted >
bosTau7_genomicSuperDups_SimFormat_AllCol.sorted
gunzip Btau_4.6.1.remap.all.germline.ucsc.gvf.gz; mv Btau_4.6.1.remap.all.germline.ucsc.gvf bosTau7_Btau_4.6.1.remap.all.germline.ucsc.gvf
grep -v ^#
bosTau7_Btau_4.6.1.remap.all.germline.ucsc.gvf | sed 's/ID=//' | sed
's/;Name/\t/' | sed 's/copy_number_/CN/’ | sed 's/variation/var/’ | awk '{print
$9”:”$3"\t"$1"\t+\t"$4"\t"$5"\t \t \t \t \t
\t \t "}' > bosTau7_dgv_SimFormat_AllCol.sorted
Download susScr3 Pig definition
files by pasting these commands:
cd GeneRef
wget http://hgdownload.cse.ucsc.edu/goldenPath/susScr3/database/refGene.txt.gz
wget http://hgdownload.cse.ucsc.edu/goldenPath/susScr3/database/refLink.txt.gz
###wget http://hgdownload.cse.ucsc.edu/goldenPath/susScr3/database/gc5BaseBw.txt.gz###
just has path below
wget http://hgdownload.cse.ucsc.edu/gbdb/susScr3/bbi/gc5Base.bw
###Use hg19 Pathways files by
querying TOUPPER
of all genes
wget http://hgdownload.cse.ucsc.edu/goldenPath/susScr3/database/cytoBandIdeo.txt.gz
wget
http://www.nature.com/nature/journal/v491/n7424/extref/nature11622-s3.xls ### xls2csv and xls2txt could work but just include in downloads <susScr3_PigSegDups_GroenenMAetalNature2012SupTable16.txt>
wget
ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/data/Sus_scrofa/by_assembly/Sscrofa3/gvf/Sscrofa3.submitted.all.germline.ucsc.gvf.gz
### < ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/data/Sus_scrofa/by_assembly/Sscrofa10.2/gvf/Sscrofa10.2.remap.all.germline.ucsc.gvf.gz
>
gunzip refGene.txt.gz; mv refGene.txt
susScr3_refGene.txt
gunzip refLink.txt.gz; mv refLink.txt
susScr3_refLink.txt
###gunzip gc5BaseBw.txt.gz; mv gc5BaseBw.txt
susScr3_gc5BaseBw.txt
perl PolishRefGeneForScanRegion_KeepOnlyChrLines.pl
susScr3_refGene.txt ### to delete chromosome “GL892101-1”...”GL896661-1” and
“JH118404-1”...”JH118996-1” (or add “chr” but input not accepted)
perl MakeExonScanRegionDefFile_refGene.pl
susScr3_refGene.txt_OnlyChr.txt
perl MakeExonScanRegionDefFile_wGene_refGene.pl
susScr3_refGene.txt_OnlyChr.txt
#cp PerlModules/scan_region.pl
.
wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/bigWigToBedGraph
chmod 777 bigWigToBedGraph
###
These steps will take considerable compute time
wget http://hgdownload.cse.ucsc.edu/gbdb/susScr3/bbi/gc5Base.bw
mv gc5Base.bw
susScr3_gc5Base.bw
./bigWigToBedGraph susScr3_gc5Base.bw
susScr3_gc5Base.bedGraph
perl Polishgc5BaseForScanRegion_KeepOnlyChrLines_5120Increments.pl
susScr3_gc5Base.bedGraph
awk -F"\t"
'{print $4"\t"$1"\t+\t"$2"\t"$3"\t \t \t \t
\t \t \t "}' susScr3_gc5Base.bedGraph_OnlyChr_5120Intervals.txt >
susScr3_gc5Base_SimFormat_AllCol.sorted ### Now unique values, rand() was added
before to $4 GC to allow duplicates in scan region since original 5 base pair
interval values are only 20,40,60,80
###perl Polishgc5BaseForScanRegion_KeepOnlyChrLines.pl
susScr3_gc5Base_SimFormat_AllCol.sorted_randDec
rm susScr3_gc5Base.bw; rm susScr3_gc5Base.bedGraph; rm susScr3_gc5Base.bedGraph_OnlyChr_5120Intervals.txt
gunzip cytoBandIdeo.txt.gz; mv cytoBandIdeo.txt susScr3_cytoBandIdeo.txt
awk -F"\t" '{print
$1$4"\t"$1"\t+\t"$2"\t"$3"\t \t \t \t \t \t
\t "}' susScr3_cytoBandIdeo.txt | sed 's/chr//' |
grep chr > susScr3_cytoBand_SimFormat_AllCol.sorted
grep -v ^# susScr3_PigSegDups_GroenenMAetalNature2012SupTable16.txt
| sort -k 1,1 -k 2,2n > susScr3_PigSegDups_GroenenMAetalNature2012SupTable16.txt.sorted #Sort By Chr Pos
awk '{print
$1”:”$2"\tchr"$1"\t+\t"$2"\t"$3"\t \t \t \t
\t \t \t "}' susScr3_PigSegDups_GroenenMAetalNature2012SupTable16.txt.sorted >
susScr3_genomicSuperDups_SimFormat_AllCol.sorted
gunzip Sscrofa3.submitted.all.germline.ucsc.gvf.gz; mv Sscrofa3.submitted.all.germline.ucsc.gvf
susScr3_Sscrofa3.submitted.all.germline.ucsc.gvf
grep -v ^# susScr3_Sscrofa3.submitted.all.germline.ucsc.gvf
| sed 's/ID=//' | sed 's/;Name/\t/' | sed 's/copy_number_/CN/’ | sed
's/variation/var/’ | sort -k 1,1 -k 4,4n | awk '{print $9”:”$3"\t"$1"\t+\t"$4"\t"$5"\t
\t \t \t \t \t \t "}' > susScr3_dgv_SimFormat_AllCol.sorted
Download dm6 Fly definition files by
pasting these commands:
cd GeneRef
wget
http://hgdownload.cse.ucsc.edu/goldenPath/dm6/database/refGene.txt.gz
wget http://hgdownload.cse.ucsc.edu/goldenPath/dm6/database/refLink.txt.gz
###wget
http://hgdownload.cse.ucsc.edu/goldenPath/dm6/database/gc5BaseBw.txt.gz
wget
http://hgdownload.cse.ucsc.edu/gbdb/dm6/bbi/gc5BaseBw/gc5Base.bw
###Use hg19 Pathways files by querying
TOUPPER of all genes
wget
http://hgdownload.cse.ucsc.edu/goldenPath/dm6/database/cytoBandIdeo.txt.gz
wget
http://humanparalogy.gs.washington.edu/dm3/data/dm3genomicSuperDup.tab ###Needs
Liftover as Instructed Below###NoHeader SeeHeader in http://paralogy.gs.washington.edu/build37/data/GRCh37GenomicSuperDup.tab
wget
ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/data/Drosophila_melanogaster/by_assembly/Release_6_plus_ISO1_MT/gvf/Release_6_plus_ISO1_MT.remap.all.germline.ucsc.gvf.gz
gunzip refGene.txt.gz; mv refGene.txt
dm6_refGene.txt
gunzip refLink.txt.gz; mv refLink.txt
dm6_refLink.txt
###gunzip gc5BaseBw.txt.gz; mv gc5BaseBw.txt
dm6_gc5BaseBw.txt
cp
../PolishRefGeneForScanRegion_KeepOnlyChrLines.pl .
perl
PolishRefGeneForScanRegion_KeepOnlyChrLines.pl dm6_refGene.txt ### to delete
non “chr” lines
perl MakeExonScanRegionDefFile_refGene.pl
dm6_refGene.txt_OnlyChr.txt
perl
MakeExonScanRegionDefFile_wGene_refGene.pl dm6_refGene.txt_OnlyChr.txt
#cp PerlModules/scan_region.pl .
wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/bigWigToBedGraph
chmod 777 bigWigToBedGraph
### These steps will take considerable
compute time
mv gc5Base.bw dm6_gc5Base.bw
./bigWigToBedGraph dm6_gc5Base.bw
dm6_gc5Base.bedGraph
cp
../Polishgc5BaseForScanRegion_KeepOnlyChrLines_5120Increments.pl .
perl
Polishgc5BaseForScanRegion_KeepOnlyChrLines_5120Increments.pl
dm6_gc5Base.bedGraph
### Illegal division by zero at
Polishgc5BaseForScanRegion_KeepOnlyChrLines_5120Increments.pl line 30,
<SAMPSHEET> line 358024431.# so no Y
awk -F"\t" '{print
$4"\t"$1"\t+\t"$2"\t"$3"\t \t \t \t \t \t \t
"}' dm6_gc5Base.bedGraph_OnlyChr_5120Intervals.txt >
dm6_gc5Base_SimFormat_AllCol.sorted ### Now unique values, rand() was added
before to $4 GC to allow duplicates in scan region since original 5 base pair
interval values are only 20,40,60,80
###perl
Polishgc5BaseForScanRegion_KeepOnlyChrLines.pl
dm6_gc5Base_SimFormat_AllCol.sorted_randDec
rm dm6_gc5Base.bw; rm dm6_gc5Base.bedGraph;
rm dm6_gc5Base.bedGraph_OnlyChr_5120Intervals.txt
gunzip
cytoBandIdeo.txt.gz; mv cytoBandIdeo.txt dm6_cytoBandIdeo.txt
awk -F"\t" '{print
$1$4"\t"$1"\t+\t"$2"\t"$3"\t \t \t \t \t \t
\t "}' dm6_cytoBandIdeo.txt | sed 's/chr//' >
dm6_cytoBand_SimFormat_AllCol.sorted
awk '{print
$1"\t"$2"\t"$3"\t"$6"#"$7"#"$8}'
dm3genomicSuperDup.tab > dm3genomicSuperDup.tab_ForLiftover.txt
### Go to
https://genome.ucsc.edu/cgi-bin/hgLiftOver Original Assembly: dm3 New Assembly:
dm6 Click Choose File Click Upload File Click View Conversions
mv hglft_genome_*.bed dm6_genomicSuperDup.tab
sort -k 1,1 -k 2,2n dm6_genomicSuperDup.tab |
sed 's/#/\t/g' > dm6_genomicSuperDup.tab.sorted #Sort By Chr Pos
awk '{print
$5":"$6"\t"$1"\t"$4"\t"$2"\t"$3"\t
\t \t \t \t \t \t "}' dm6_genomicSuperDup.tab.sorted >
dm6_genomicSuperDups_SimFormat_AllCol.sorted
gunzip Release_6_plus_ISO1_MT.remap.all.germline.ucsc.gvf.gz;
mv Release_6_plus_ISO1_MT.remap.all.germline.ucsc.gvf
dm6_Release_6_plus_ISO1_MT.remap.all.germline.ucsc.gvf
grep -v ^#
dm6_Release_6_plus_ISO1_MT.remap.all.germline.ucsc.gvf | sed 's/ID=//' | sed
's/;Name/\t/' | sed 's/copy_number_/CN/' | sed 's/variation/var/' | awk '{print
$9":"$3"\t"$1"\t+\t"$4"\t"$5"\t \t
\t \t \t \t \t "}' > dm6_dgv_SimFormat_AllCol.sorted
cp hg19_bioCycPathway.txt
dm6_bioCycPathway.txt
cp hg19_cgapAlias.txt dm6_cgapAlias.txt
cp hg19_cgapBiocDesc.txt dm6_cgapBiocDesc.txt
cp hg19_cgapBiocPathway.txt
dm6_cgapBiocPathway.txt
cp hg19_keggMapDesc.txt dm6_keggMapDesc.txt
cp hg19_keggPathway.txt dm6_keggPathway.txt
Download canFam2 Dog definition
files by pasting these commands:
cd GeneRef
wget
http://hgdownload.cse.ucsc.edu/goldenPath/canFam2/database/refGene.txt.gz
wget
http://hgdownload.cse.ucsc.edu/goldenPath/canFam2/database/refLink.txt.gz
wget
http://hgdownload.cse.ucsc.edu/goldenPath/canFam2/database/gc5Base.txt.gz
###Use hg19 Pathways files by querying
TOUPPER of all genes
###No Cytobands! wget
http://hgdownload.cse.ucsc.edu/goldenPath/canFam2/database/cytoBandIdeo.txt.gz
wget
http://humanparalogy.gs.washington.edu/canFam2seqdup/canFam2_wgac
wget
ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/data/Canis_lupus/by_assembly/CanFam2.0/gvf/CanFam2.0.submitted.all.germline.ucsc.gvf.gz
gunzip refGene.txt.gz; mv refGene.txt
canFam2_refGene.txt
gunzip refLink.txt.gz; mv refLink.txt
canFam2_refLink.txt
###gunzip gc5BaseBw.txt.gz; mv gc5BaseBw.txt canFam2_gc5BaseBw.txt
cp
../PolishRefGeneForScanRegion_KeepOnlyChrLines.pl .
perl
PolishRefGeneForScanRegion_KeepOnlyChrLines.pl canFam2_refGene.txt ### to
delete non “chr” lines
perl MakeExonScanRegionDefFile_refGene.pl
canFam2_refGene.txt_OnlyChr.txt
perl
MakeExonScanRegionDefFile_wGene_refGene.pl canFam2_refGene.txt_OnlyChr.txt
#cp PerlModules/scan_region.pl .
gunzip gc5Base.txt.gz; mv gc5Base.txt
canFam2_gc5Base.txt
awk -F"\t" '{print $1"\t"$2"\t+\t"$3"\t"$4"\t
\t \t \t \t \t \t "}' canFam2_gc5Base.txt >
canFam2_gc5Base_SimFormat_AllCol.sorted ### Now unique values, rand() was added
before to $4 GC to allow duplicates in scan region since original 5 base pair
interval values are only 20,40,60,80
###perl
Polishgc5BaseForScanRegion_KeepOnlyChrLines.pl canFam2_gc5Base_SimFormat_AllCol.sorted_randDec
rm canFam2_gc5Base.txt
sort -k 1,1 -k 2,2n canFam2_wgac | grep -v chrom
> canFam2_genomicSuperDup.tab.sorted #Sort By Chr Pos
awk '{print $4":"$5"\t"$1"\t+\t"$2"\t"$3"\t
\t \t \t \t \t \t "}' canFam2_genomicSuperDup.tab.sorted >
canFam2_genomicSuperDups_SimFormat_AllCol.sorted
gunzip
CanFam2.0.submitted.all.germline.ucsc.gvf.gz; mv
CanFam2.0.submitted.all.germline.ucsc.gvf
canFam2_CanFam2.0.submitted.all.germline.ucsc.gvf
grep -v ^# canFam2_CanFam2.0.submitted.all.germline.ucsc.gvf
| sed 's/ID=//' | sed 's/;Name/\t/' | sed 's/copy_number_/CN/' | sed
's/variation/var/' | awk '{print
$9":"$3"\t"$1"\t+\t"$4"\t"$5"\t \t
\t \t \t \t \t "}' > canFam2_dgv_SimFormat_AllCol.sorted
cp hg19_bioCycPathway.txt canFam2_bioCycPathway.txt
cp hg19_cgapAlias.txt canFam2_cgapAlias.txt
cp hg19_cgapBiocDesc.txt canFam2_cgapBiocDesc.txt
cp hg19_cgapBiocPathway.txt canFam2_cgapBiocPathway.txt
cp hg19_keggMapDesc.txt canFam2_keggMapDesc.txt
cp hg19_keggPathway.txt canFam2_keggPathway.txt
cp hg19_cytoBand_SimFormat_AllCol.sorted
canFam2_cytoBand_SimFormat_AllCol.sorted
cp hg19_cytoBand.txt canFam2_cytoBandIdeo.txt
Download oviAri3 Sheep definition
files by pasting these commands:
cd GeneRef
wget http://hgdownload.cse.ucsc.edu/goldenPath/oviAri3/database/refGene.txt.gz
wget
http://hgdownload.cse.ucsc.edu/goldenPath/oviAri3/database/refFlat.txt.gz
wget
http://hgdownload.cse.ucsc.edu/gbdb/oviAri3/bbi/gc5Base.bw #Takes awhile since
a huge file
wget http://hgdownload.cse.ucsc.edu/goldenPath/oviAri3/database/cytoBandIdeo.txt.gz
wget
https://static-content.springer.com/esm/art%3A10.1186%2Fs12864-017-3690-x/MediaObjects/12864_2017_3690_MOESM1_ESM.xlsx
#Table S1.6 copy coordinates into text file oviAri3_genomicSuperDups_Feng2017.txt
wget
ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/archive/old_format/Ovis_aries/by_assembly/Oar_v3.1/gvf/Oar_v3.1.submitted.all.germline.ucsc.gvf.gz
gunzip refGene.txt.gz; mv refGene.txt
oviAri3_refGene.txt
gunzip refFlat.txt.gz; mv refFlat.txt
oviAri3_refLink.txt
awk '{ORS="";print
$1"\t"$2"\t"$2"\t";print;print"\n"}'
oviAri3_refLink.txt > a; mv oviAri3_refLink.txt oviAri3_refLink.txtORIG; mv
a oviAri3_refLink.txt
perl
PolishRefGeneForScanRegion_KeepOnlyChrLines.pl oviAri3_refGene.txt ### to
delete non chr lines
perl MakeExonScanRegionDefFile_refGene.pl
oviAri3_refGene.txt_OnlyChr.txt
perl
MakeExonScanRegionDefFile_wGene_refGene.pl oviAri3_refGene.txt_OnlyChr.txt
wget
hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/bigWigToBedGraph
chmod 777 bigWigToBedGraph
mv gc5Base.bw oviAri3_gc5Base.bw
./bigWigToBedGraph oviAri3_gc5Base.bw
oviAri3_gc5Base.bedGraph #Takes awhile since a huge file
perl
Polishgc5BaseForScanRegion_KeepOnlyChrLines_5120Increments.pl
oviAri3_gc5Base.bedGraph #Takes awhile since a huge file
awk -F"\t" '{print
$4"\t"$1"\t+\t"$2"\t"$3"\t \t \t \t \t \t \t
"}' oviAri3_gc5Base.bedGraph_OnlyChr_5120Intervals.txt >
oviAri3_gc5Base_SimFormat_AllCol.sorted ### Now unique values, rand() was added
before to $4 GC to allow duplicates in scan region since original 5 base pair
interval values are only 20,40,60,80
rm oviAri3_gc5Base.bw; rm
oviAri3_gc5Base.bedGraph; rm oviAri3_gc5Base.bedGraph_OnlyChr_5120Intervals.txt
gunzip cytoBandIdeo.txt.gz; mv
cytoBandIdeo.txt oviAri3_cytoBandIdeo.txt
awk -F"\t" '{print
$1$4"\t"$1"\t+\t"$2"\t"$3"\t \t \t \t \t \t
\t "}' oviAri3_cytoBandIdeo.txt | sed 's/chr//' >
oviAri3_cytoBand_SimFormat_AllCol.sorted
mv oviAri3_genomicSuperDups_Feng2017.txt
oviAri3_GenomicSuperDupgenomicSuperDup.tab
sort -k 1,1 -k 2,2n
oviAri3_GenomicSuperDupgenomicSuperDup.tab >
oviAri3_GenomicSuperDupgenomicSuperDup.tab.sorted #Sort By Chr Pos
awk '{print
NR"\tchr"$1"\t+\t"$2"\t"$3"\t \t \t \t \t \t
\t "}' oviAri3_GenomicSuperDupgenomicSuperDup.tab.sorted >
oviAri3_genomicSuperDups_SimFormat_AllCol.sorted
gunzip
Oar_v3.1.submitted.all.germline.ucsc.gvf.gz; mv
Oar_v3.1.submitted.all.germline.ucsc.gvf
oviAri3_Oar_v3.1.submitted.all.germline.ucsc.gvf
grep -v ^#
oviAri3_Oar_v3.1.submitted.all.germline.ucsc.gvf | sed 's/ID=//' | sed 's/;Name/\t/'
| sed 's/copy_number_/CN/' | sed 's/variation/var/' | awk '{print
$9":"$3"\t"$1"\t+\t"$4"\t"$5"\t \t
\t \t \t \t \t "}' > oviAri3_dgv_SimFormat_AllCol.sorted
TestConcordance.pl compares pairs of CNV calls.
The pair can be PennCNV vs. Nexus of the same
sample, Mother vs. Child, Tumor vs. Blood, Transplant
Donor vs. Recipient, technical replicate 1 vs. technical replicate 2.
The input is calls in PennCNV format with confidence
column and Algorithm column, sorted by ID and algorithm. You also need to
provide a map file for all probes being considered. Make sure the map is sorted
by position and chromosome (sort -n -k 4,4 file.map | sort -s > file_Sorted.map).
Multiple CNV Detection Comparison
and De Novo CNV Calls Annotation
ArrayConcordance_InheritanceDenovo_AnnotateCNVCalls
Folder Scripts
COMMAND:
perl PQN_Concord_Inh.pl Cases_Denovo.rawcnv
Cases_Denovo_Q.rawcnv Cases_Denovo_N.rawcnv Cases_Denovo.fam ChrSnp0Pos.map
--cluster
You need a parallel compute cluster you can qsub to
use --cluster option, otherwise run without this option to run in serial
fashion.
INPUTS:
Make sure to enter a blank line at the end.
Cases_Denovo.rawcnv
chr1:1-3
numsnp=3 length=3
state1,cn=0 Mom.baflrr startsnp=rs1 endsnp=rs3 conf=10 PennCNV
chr1:4-7
numsnp=4 length=4
state5,cn=3 Dad.baflrr startsnp=rs4 endsnp=rs7 conf=10 PennCNV
chr1:1-3
numsnp=3 length=3
state1,cn=0 Pro.baflrr startsnp=rs1 endsnp=rs3 conf=10 PennCNV
chr1:4-7
numsnp=4 length=4
state5,cn=3 Pro.baflrr startsnp=rs4 endsnp=rs7 conf=10 PennCNV
chr1:8-10 numsnp=3 length=3 state5,cn=3 Pro.baflrr startsnp=rs8
endsnp=rs10 conf=10 PennCNV
Cases_Denovo_Q.rawcnv
chr1:1-3
numsnp=3 length=3
state1,cn=0 Mom.baflrr startsnp=rs1 endsnp=rs3 conf=10 QuantiSNP
chr1:4-7
numsnp=4 length=4 state5,cn=3
Dad.baflrr startsnp=rs4 endsnp=rs7 conf=10 QuantiSNP
chr1:1-3
numsnp=3 length=3
state1,cn=0 Pro.baflrr startsnp=rs1 endsnp=rs3 conf=10 QuantiSNP
chr1:4-7
numsnp=4 length=4
state5,cn=3 Pro.baflrr startsnp=rs4 endsnp=rs7 conf=10 QuantiSNP
chr1:8-9
numsnp=2 length=2
state5,cn=3 Pro.baflrr startsnp=rs8 endsnp=rs9 conf=10 QuantiSNP
Cases_Denovo_N.rawcnv
chr1:1-3
numsnp=3 length=3
state1,cn=0 Mom.baflrr startsnp=rs1 endsnp=rs3 conf=10 Nexus
chr1:4-7
numsnp=4 length=4
state5,cn=3 Dad.baflrr startsnp=rs4 endsnp=rs7 conf=10 Nexus
chr1:1-3
numsnp=3 length=3
state1,cn=0 Pro.baflrr startsnp=rs1 endsnp=rs3 conf=10 Nexus
chr1:4-7
numsnp=4 length=4
state5,cn=3 Pro.baflrr startsnp=rs4 endsnp=rs7 conf=10 Nexus
chr1:8-9
numsnp=2 length=2
state5,cn=3 Pro.baflrr startsnp=rs8 endsnp=rs9 conf=10 Nexus
Cases_Denovo.fam
FID IID FatID MotID Gend Aff
1 Mom.baflrr 0 0 0 1
1 Dad.baflrr 0 0 0 1
1 Pro.baflrr Dad.baflrr Mom.baflrr 0 2
ChrSnp0Pos.map
Chr Snp Cm Pos
1 rs1 0 1
1 rs2 0 2
1 rs3 0 3
1 rs4 0 4
1 rs5 0 5
1 rs6 0 6
1 rs7 0 7
1 rs8 0 8
1 rs9 0 9
1 rs10 0 10
OUTPUTS:
The
program does not currently generate new overlapping
segments consensus CNV calls from multiple algorithm comparison for further analysis.
I have used PennCNV calls that had good overlap in QuantiSNP rather than
generating an overlapping segments consensus call. Many CNV calling algorithms
are conservative in the boundary calls, especially if you did not run the
PennCNV clean_cnv.pl script to merge fragmented CNVs.
Make sure you look at either PvsQ or QvsP CallMatching and MatchSummary, looking at both will be confusing. For the QvsP output, the first 9 columns refer to the QuantiSNP calls. The next 6 columns refer to the overlap profile of the QuantiSNP call with the PennCNV call. The next 9 columns starting with RefCallMatchingCN_CNVBoundariesChrStartStop refer to the overlapping PennCNV call with the same CN contributing to the “hitsnp” column. The next 9 columns starting with RefCallDiffCN_CNVBoundariesChrStartStop refer to the overlapping PennCNV call with a different CN contributing to the “hitDiffCN” column.
The columns with numbers in the QvsP.txtCNVMatchSummary.txt file are the hitDiffCN mismatches. For example, 01 is the count of QuaniSNP calls that were CN=0 but PennCNV called as CN=1.
PennCNV_ConcordInh_NoROH.txt
We see full overlap of sample level rawcnv calls of
the three algorithms PennCNV, QuanitSNP, and Nexus with reference to the
PennCNV calls (indicated by “vsP”), except PennCNV chr1:8-10 where the other
algorithms called chr1:8-9 but this
still is a consistently detected CNV.
We see full inheritance reporting with inheritance
from mother chr1:1-3, inheritance from father chr1:4-7, and de novo chr1:8-10
for the proband CNV calls. We see inheritance to proband chr1:1-3 for the
mother CNV call. We see inheritance to proband chr1:4-7 for the father CNV
call. In the case of a CNV overlapping but with a different CN state between
family members, similar columns are reported with an additional column
specifying the different CN state. Certainly CN 3 and 4 seems the most typical
permissible different CN to allow as a match but others may indicate abnormal
noise patterns.
|
CNV Boundaries Chr StartStop
Hg19 |
numsnp |
Length (bp) |
CN |
ChipID |
startsnp |
endsnp |
confidence |
algorithm |
50% PennCNV Overlap |
Freq PennCNV Overlap |
50% QuantiSNP Overlap |
Freq QuantiSNP Overlap |
50% Nexus Overlap |
Freq Nexus Overlap |
BlindedID |
Inherit From Mother 50% Overlap |
Freq Inherit From Mother Overlap |
Inherit From Father 50% Overlap |
Freq Inherit From Father Overlap |
Inherit To Proband 50% Overlap |
Freq Inherit To Proband Overlap |
Inherit From Mother 50% Overlap
Diff CN |
Freq Inherit From Mother Overlap
Diff CN |
Inherit From Mother Diff CN |
Inherit From Father 50% Overlap
Diff CN |
Freq Inherit From Father Overlap
Diff CN |
Inherit From Father Diff CN |
Inherit To Proband 50% Overlap
Diff CN |
Freq Inherit To Proband Overlap
Diff CN |
Inherit To Proband Diff CN |
MFP Trio State (D=Diploid,
2=LOH, N=NA) |
Denovo Call |
Gene |
Distance |
|
chr1:1-3 |
3 |
3 |
0 |
Pro.baflrr |
rs1 |
rs3 |
10 |
PennCNV |
Y |
1 |
Y |
1 |
Y |
1 |
Pro.baflrr |
Y |
1 |
N |
0 |
na |
na |
N |
0 |
. |
N |
0 |
. |
na |
na |
na |
0D0 |
NotDenovo |
DDX11L1,DDX11L9 |
11871 |
|
chr1:4-7 |
4 |
4 |
3 |
Pro.baflrr |
rs4 |
rs7 |
10 |
PennCNV |
Y |
1 |
Y |
1 |
Y |
1 |
Pro.baflrr |
N |
0 |
Y |
1 |
na |
na |
N |
0 |
. |
N |
0 |
. |
na |
na |
na |
D33 |
NotDenovo |
DDX11L1,DDX11L9 |
11867 |
|
chr1:8-10 |
3 |
3 |
3 |
Pro.baflrr |
rs8 |
rs10 |
10 |
PennCNV |
Y |
1 |
Y |
0.667 |
Y |
0.667 |
Pro.baflrr |
N |
0 |
N |
0 |
na |
na |
N |
0 |
. |
N |
0 |
. |
na |
na |
na |
DD3 |
Denovo |
DDX11L1,DDX11L9 |
11864 |
|
chr1:1-3 |
3 |
3 |
0 |
Mom.baflrr |
rs1 |
rs3 |
10 |
PennCNV |
Y |
1 |
Y |
1 |
Y |
1 |
Mom.baflrr |
na |
na |
na |
na |
Y |
1 |
na |
na |
na |
na |
na |
na |
N |
0 |
. |
NNN |
NotDenovo |
DDX11L1,DDX11L9 |
11871 |
|
chr1:4-7 |
4 |
4 |
3 |
Dad.baflrr |
rs4 |
rs7 |
10 |
PennCNV |
Y |
1 |
Y |
1 |
Y |
1 |
Dad.baflrr |
na |
na |
na |
na |
Y |
1 |
na |
na |
na |
na |
na |
na |
N |
0 |
. |
NNN |
NotDenovo |
DDX11L1,DDX11L9 |
11867 |
changing Cases_Denovo.rawcnv
chr1:1-3 numsnp=3
length=3 state1,cn=0 Mom.baflrr
startsnp=rs1 endsnp=rs3 conf=10 PennCNV
to
chr1:1-3
numsnp=3 length=3
state2,cn=1 Mom.baflrr startsnp=rs1 endsnp=rs3 conf=10 PennCNV
and reviewing the output PennCNV_ConcordInh_NoROH.txt
50%QuantiSNPOverlap and 50%NexusOverlap become N
since the CN state is different.
InheritFromMother50%Overlap becomes N and
InheritFromMother50%OverlapDiffCN becomes Y with InheritFromMotherDiffCN 1 for
the proband CNV call.
InheritToProband50%Overlap becomes N and
InheritToProband50%OverlapDiffCN becomes Y with InheritToProbandDiffCN 0 for
the mother CNV call.
|
CNV Boundaries Chr
StartStop Hg19 |
numsnp |
Length (bp) |
CN |
ChipID |
startsnp |
endsnp |
confidence |
algorithm |
50% PennCNV Overlap |
Freq PennCNV Overlap |
50% QuantiSNP Overlap |
Freq QuantiSNP Overlap |
50% Nexus Overlap |
Freq Nexus Overlap |
BlindedID |
Inherit From Mother 50%
Overlap |
Freq Inherit From Mother
Overlap |
Inherit From Father 50%
Overlap |
Freq Inherit From Father
Overlap |
Inherit To Proband 50%
Overlap |
Freq Inherit To Proband
Overlap |
Inherit From Mother 50%
Overlap Diff CN |
Freq Inherit From Mother
Overlap Diff CN |
Inherit From Mother Diff
CN |
Inherit From Father 50%
Overlap Diff CN |
Freq Inherit From Father
Overlap Diff CN |
Inherit From Father Diff
CN |
Inherit To Proband 50%
Overlap Diff CN |
Freq Inherit To Proband
Overlap Diff CN |
Inherit To Proband Diff CN |
MFP Trio State (D=Diploid,
2=LOH, N=NA) |
Denovo Call |
Gene |
Distance |
|
chr1:1-3 |
3 |
3 |
0 |
Pro.baflrr |
rs1 |
rs3 |
10 |
PennCNV |
Y |
1 |
Y |
1 |
Y |
1 |
Pro.baflrr |
N |
0 |
N |
0 |
na |
na |
Y |
1 |
1 |
N |
0 |
. |
na |
na |
na |
1D0 |
NotDenovo |
DDX11L1,DDX11L9 |
11871 |
|
chr1:4-7 |
4 |
4 |
3 |
Pro.baflrr |
rs4 |
rs7 |
10 |
PennCNV |
Y |
1 |
Y |
1 |
Y |
1 |
Pro.baflrr |
N |
0 |
Y |
1 |
na |
na |
N |
0 |
. |
N |
0 |
. |
na |
na |
na |
D33 |
NotDenovo |
DDX11L1,DDX11L9 |
11867 |
|
chr1:8-10 |
3 |
3 |
3 |
Pro.baflrr |
rs8 |
rs10 |
10 |
PennCNV |
Y |
1 |
Y |
0.667 |
Y |
0.667 |
Pro.baflrr |
N |
0 |
N |
0 |
na |
na |
N |
0 |
. |
N |
0 |
. |
na |
na |
na |
DD3 |
Denovo |
DDX11L1,DDX11L9 |
11864 |
|
chr1:1-3 |
3 |
3 |
1 |
Mom.baflrr |
rs1 |
rs3 |
10 |
PennCNV |
Y |
1 |
N |
0 |
N |
0 |
Mom.baflrr |
na |
na |
na |
na |
N |
0 |
na |
na |
na |
na |
na |
na |
Y |
1 |
0 |
NNN |
NotDenovo |
DDX11L1,DDX11L9 |
11871 |
|
chr1:4-7 |
4 |
4 |
3 |
Dad.baflrr |
rs4 |
rs7 |
10 |
PennCNV |
Y |
1 |
Y |
1 |
Y |
1 |
Dad.baflrr |
na |
na |
na |
na |
Y |
1 |
na |
na |
na |
na |
na |
na |
N |
0 |
. |
NNN |
NotDenovo |
DDX11L1,DDX11L9 |
11867 |
PennCNV_ConcordInh_NoROH_Summary.txt
Sample based counts
|
ChipID |
BlindedID |
Total Calls |
PennCNV Overlap |
QuantiSNP Overlap |
Nexus Overlap |
Three Overlap |
Two Overlap |
One Overlap |
Inherit From Mother |
Inherit From Father |
Inherit To Proband |
Denovo |
Inherit From Mother And Father
Same CN |
Inherit From Mother And Father
Diff CN |
MFP=110 |
MFP=334 |
Three Overlap And Inh |
Two Overlap And Inh |
One Overlap And Inh |
Three Overlap And Inh Under
Fiftykb |
Three Overlap And Inh Fifty
To Two Hundred kb |
Three Overlap And Inh Above
Two Hundred kb |
Two Overlap And Inh Under Fifty
kb |
Two Overlap And Inh Fifty To
Two Hundred kb |
Two Overlap And Inh Above Two
Hundred kb |
One Overlap And Inh Under Fifty
kb |
One Overlap And Inh Fifty To
Two Hundred kb |
One Overlap And Inh Above Two
Hundred kb |
Family Status |
|
Pro.baflrr |
Pro.baflrr |
3 |
3 |
3 |
3 |
3 |
0 |
0 |
1 |
1 |
0 |
1 |
0 |
0 |
0 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
trio |
|
Mom.baflrr |
Mom.baflrr |
1 |
1 |
1 |
1 |
1 |
0 |
0 |
0 |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
trio |
|
Dad.baflrr |
Dad.baflrr |
1 |
1 |
1 |
1 |
1 |
0 |
0 |
0 |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
trio |
|
|
|
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
duo |
|
|
|
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
single |
De novo Prioritization
Trio Recall PennCNV
At least 2 algorithms call de novo (P, Q, N) (do not
exclude parent CNV called by 1 algorithm that is inherited to child CNV called
by 2 algorithms as more de novos will result)
Parental Origin low p (if enough informative markers)
Greater than 5 SNPs
Low/absent untransmitted rates
Low/absent Control (DGV, SSC Healthy Trios, CHOP
Control, Framingham)
BAF/LRR Review (full trio in case FN Parent)
ddPCR Validation
For each study:
ParseCNV --includePed
Plink --covar .pca.evec
PennCNV scan_region SNP based .stats results cohort A→closest
SNP results cohort B
METAL to generate meta-analysis p-value SNP based
InsertPlinkPValues Input is general tab delimited P, SNP, and one of
BETA/T/OR header present.
You
can run for each study to get study specific red flag confidence scores or run
ParseCNV and Plink again on the combined data to use as input for combined
study red flags
ParseCNV generates the output *CNV_Brief.stats which
is the SNP based CNV statistics which can be used for meta-analysis using
METAL. Since you have different arrays, the intersection SNP set will be low so
I would use the gene name as the marker rather than the SNP name. Using PennCNV
scan_region.pl with input chrX:start-stop for each SNP will annotate the closest
gene to each SNP. Then sort -g in unix to sort by p-value so best (lowest)
p-value is first since that is the one METAL will use.
run ParseCNV for each study to generate the p-values
for each study separately.
You need to rearrange *CNV_Brief.stats into a
scan_region.pl input using:
awk '{print
"chr"$2":"$3"-"$3}' CNV_Brief.stats | sed '1d'
or more specifically with all the other columns:
awk '{print
"chr"$2":"$3"-"$3"\t"$1"\t"$4"\t"$5"\t"$6"\t"$7"\t"$8"\t"$9"\t"$10"\t"$11}'
CNV_Brief.stats | sed '1d'
scan_region.pl wants no header (sed '1d') while METAL
needs the header.
The analysis looks like this:
./metal
SEPARATOR
TAB
MARKER Gene
EFFECT log(ORDel)
PVAL DelTwoTailed
PROCESS File1_ForScanRegion_GeneExon_SortDelP.txt
PROCESS File2_ForScanRegion_GeneExon_SortDelP.txt
....
ANALYZE
and again for Dups:
EFFECT log(ORDup)
PVAL DupTwoTailed
PROCESS File1_ForScanRegion_GeneExon_SortDupP.txt
PROCESS File2_ForScanRegion_GeneExon_SortDupP.txt
....
ANALYZE
Convert CNV Calls Formats from
Different Algorithms
PennCNV is the motivating input. The PennCNV input is
very human readable, widely used and informative. CNV .vcf is widely used in
sequencing applications, but not human readable. Therefore, the idea is to
convert CNVs called by different algorithms into PennCNV format with a final
column stating the algorithm for tracking. In most cases, the CNV calls file
columns and coding can be edited relatively easily in Excel. The main exception
is BirdSuite and GenomeStrip .vcf matrix formats. For these common yet very
different formats, use ConvertVCFToPennCNV.pl. For the commonly used QuantiSNP
conversion use ConvertQuantiSNPToPennCNV.pl.
PennCNV, QuantiSNP, Nexus, IlluminaGenomeStudio,
TCGA, AgilentGenomicWorkbench/Cytogenomics, AffymetrixGenotypingConsole, BirdSuite,
CGHscape, NimbleScan, R-GADA, Conifer, XHMM, Plink .cnv file cnvPartition,
GenomeStrip .vcf DEL DUP INS INV CNV
Similar settings should be used between algorithms to
ensure comparability such as using GC base content correction.
Nexus “Min Region” should be used not “Chromosome
Region” since that is the average position between probes. AgilentGenomicWorkbench/Cytogenomics
has the sample ID listed only once as a header of the CNV calls subsections.
ConvertQuantiSNPToPennCNV.pl
ConvertVCFToPennCNV_XHMM.pl
ConvertVCFToPennCNV_GenomeStrip.pl
Consider CallRate, LRR_SD, GCWF, CountCNV, PCA, and
PI_HAT as the most important sample quality metrics. CallRate and LRR_SD are
widely used and critical to evaluate. The problem is these metrics are derived
from various sources, even if you use SNP arrays with PennCNV, only LRR_SD and
GCWF are provided in the PennCNV .log. CountCNV is automatically figured out by
the .rawcnv files. CallRate, PCA, and PI_HAT are based on genotyping A/T/C/G,
not CNVs. For CallRate, you can provide Plink --missing .imiss, Illumina LIMs
Project Detail Report .csv, or Illumina Genome Studio Samples Table. PCA
population stratification components can be Eigenstrat smartPCA .pca.evec or
Plink .mds. PI_HAT is from Plink --genome .genome file. PCA is not
automatically used to create IDs to remove, which would be done for a study
where almost all samples are tightly clustered and a few outliers are then
removed. Typically the PCA shows admixture and various ethnicities and
population stratification correction without sample removal based on PCA is the
best approach.
For Illumina 550k data, key data quality metric
thresholds we have observed are: call rate > 98%, SD LRR < 0.3, |GCWF|
< 0.05, and count CNV< 100.
For Affymetrix 6.0 data, these measures include: call
rate > 96%, SD LRR < 0.35, |GCWF| < 0.02, and count CNV < 80.
Inputs: (all are recommended but not
required to run ParseCNV_QC.pl)
PennCNV .log
PennCNV .rawcnv
Plink --missing .imiss / Illumina LIMs Project Detail Report .csv /
Illumina Genome Studio Samples Table / General format: SampleID CallRate
Eigenstrat smartPCA .pca.evec / Plink .mds
Plink --genome .genome
perl ParseCNV_QC.pl --log PennCNV_Omni25.log --rawcnv
file.rawcnv --callrate PCGC_Omni25-8v1_BED_Miss.imiss --popstrat file.pca.evec
--related PCGC_Omni25-8v1_BED_GenomeAll.genome
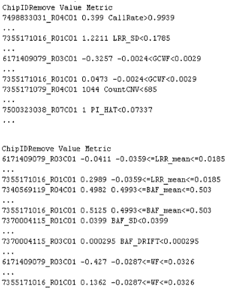
Outputs:
See QC_Plot.pdf sample QC distributions with outliers
to remove in blue and corresponding QC_RemoveIDs.txt with ChipIDs failing the
various QC metrics. You may delete some rows from this file if you want a more
aggressive ChipID/Sample inclusion or if the threshold does not look correct on
the plot. Remove thresholds for each quality metric are determined by the R
package extremevalues function getOutliers. You can also use exclusions based
on other PennCNV log metrics provided in QC_Plot_2.pdf and QC_RemoveIDs_LRRmean_BAFmean_BAFSD_BAFDRIFT_WF.txt.
Then run this code found in the GeneRef folder to
create a "clean" .rawcnv file to use for ParseCNV association
perl FilterCNV.pl QC_RemoveIDs.txt CNVCalls.rawcnv 5
remove
UPDATE: The R getOutliers is not very dynamic in
selecting outliers based on a variety of possible data quality metric
distributions. This typically results in too many samples being excluded.
Again, look for linear mode for inclusion and exponential mode for exclusion.
This can be easily done in R or Excel. Once an appropriate threshold is
determined from reviewing the first run distributions or from a static know
threshold for consistency, ParseCNV_QC.pl can be run again specifying
thresholds to drive the plots and sample remove report.
Run once with no
thresholds specified and review automatically determined thresholds based on
outliers in your dataset.
Then you may
specify some adjusted threshold for a given quality metric based on your review
of the plots.
For example add: --qccallrate 0.98 --qclrrsd 0.2 --qcgcwf
0.005 –qcnumcnv 100
popstrat is just for plotting, no exclusion is
currently supported based on popstrat.
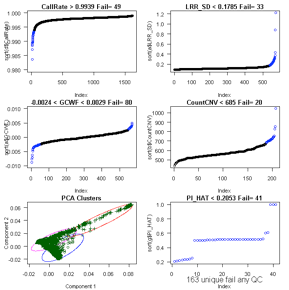
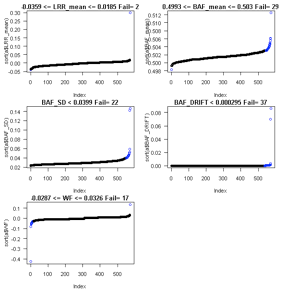
Example: running ParseCNV_QC again after reviewing
plots. See 146 vs. 49 samples excluded based on automatic vs. manual quality
metric thresholds (see bottom of plot “unique fail any QC”):
perl ParseCNV_QC_3_ProvideFilterThresholds.pl --log
PennCNV_Omni1.log --rawcnv file.rawcnv --callrate
PCGC_Omni25-8v1_BED_Miss.imiss --popstrat file.pca.evec
perl ParseCNV_QC_3_ProvideFilterThresholds.pl --log
PennCNV_Omni1.log --rawcnv file.rawcnv --callrate
PCGC_Omni25-8v1_BED_Miss.imiss --popstrat file.pca.evec --qcnumcnv 700 --qcgcwf 0.005 --qclrrsd
0.2 --qccallrate 0.98
Bimodal metrics indicate batches, typically of different
chip versions
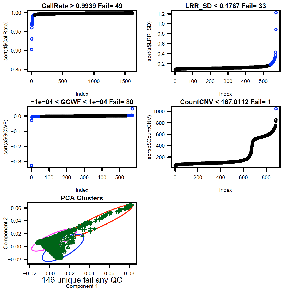
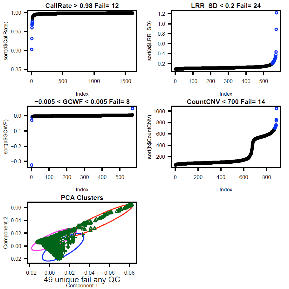
CNV Call Quality Control:
Removing CNV calls may introduce significant bias and
there are a limited number of metrics to make an informed decision. Probably
the best metric is the confidence score based on the cumulative probability
from the HMM. ParseCNV annotates average numsnp, length, and confidence of each
potential CNVR association which captures this quality/confidence issue with
less bias than upfront CNV call quality control.
Step by Step Processing Description:
CNV calls are mapped into SNP based statistics which are
then merged into CNVRs based on proximity (1Mb) and similar significance (power
of ten P value) of neighboring SNPs.
The most significant subregion is presented in the
case of multiple significant proximal CNVRs.
Deletion and Duplication p values are then calculated
by pooling 0 and 1 copy for deletion and 3 and 4 for duplication.
The runtime log lists the processing
steps:
scan_range2: Takes SNP based statistics and
collapses into CNVRs based on distance 1Mb and p-value one power of ten in
negative log.
scan_region: Takes CNVR and annotates nearest
UCSC gene (more inclusive than RefSeq genes) and proximal distance until gene
boundary (including introns, 0 is direct overlap). Definition files can be
updated or different builds used by downloading from UCSC Tables.
scan_DescPathway: Takes CNVR with gene and gives full
text description of gene name and pathway (many not_found in current version of
definition file).
scan_rangeJustPos2: Takes SNP based statistics and
gives SNP indexes based only on distance 1MB to limit redundancy of small
regions with different significance levels.
CNVToPed: --includePed since large file. Del
and Dup Ped files created for more statistics in Plink. In the Del Ped,
state1cn=0 → 1 1 and state2cn=1 → 1 2 other → 2 2. In the Dup
Ped, state5cn=3 → 1 2 state6cn=4 → 1 1 other → 2 2. This definition
closely resembles the CNV state frequencies (1 1=rare, 1 2=common, 2 2=very
common). Note many probes will be always diploid (no CNV calls) so NA will
result in Plink. Sorted.map created automatically should be used with the
resulting ped.
vlookup SNP to
Region ID: Add
column Region ID.
countBarcodeOCCURENCE_V2: Count Region ID occurrences.
vlookup Count Sig
Regions: Add
column Count Region ID.
Sort by p-value: Sort from Chr-Pos to low->high p-value.
Data-filter-advanced-unique
records only:
Include the first occurrence of each Region ID to filter out many significant
regions close together, keeping the lowest p-value occurrence.
Create UCSC Custom
Track For Review:
UCSCGenomeBrowser-Select Assembly-add custom tracks-Browse-Submit
NUMLines: Adds Tab delimited Index Column to
space delimited CNV calls (.rawcnv) files to access recorded contributing calls
to each association for back referencing trackability
ComponentDelCallsCases,ComponentDupCallsCases,ComponentDelCallsControls,ComponentDupCallsControls
PercentSamples-AverageNumsnpLenComponentCalls: Adds AverageNumsnps AverageLength
CNVRange for cases and controls for screening purposes
SpecificBafLrrAccessMany: Assumes all BafLrr files have
ProbeID as first column and have same probe sorted error, but can be different
from map. If Probe ID not first column, provide probe order reference file
--probesBafLrr <file.txt>. Needs additional SampleID to BafLrrFilePath
file specified on command line by --idToPath <file.txt>. Highly
recommended if BafLrr files available. Takes CNVRs and contributing sample IDs
and creates files with BAF and LRR values for each CNVR with all samples and a
master file with all CNVRs and signals in all samples with CNV contributing to
association in BafLrr Folder. Used for review of specific association region
across many samples.
Main output: CNVR_ALL_ReviewedCNVRs.txt Note results sorted by deletion p are
followed by results sorted by duplication p (See CNVType column).
Copy and paste text into
EXCEL to review output! Do not try viewing in shell terminal except perhaps
*_brief.txt.
For
big data, cells may overflow to the next line since the total number of
characters that a cell can contain is 32,767 characters. The Sample
ID columns are usually the problem. You will notice this problem if the first
column is not all chr:start-stop values. To fix this, add a column with an
index, sort by the chr:start-stop column, delete rows not starting with “chr”
at the top and bottom, and sort by the index. Full details will not be present
for very common CNVs but these are typically not of interest anyway. Also
consider making Sample IDs a simple index number rather than a lengthy text
string. You can use vlookup in Excel to replace sample IDs with index sample
IDs in your input .rawcnv file.
To view output files in OpenOfficeCalc (Linux vs Dos
endlines could cause blank lines):
RightClick-OpenWith-OpenOffice.orgCalc-TextImport=Defaults
If blank lines: SelectRowsNotHeader-Data-Filter-StandardFilter-Value=Empty-RightClick-Delete
Rows-Data-Filter-RemoveFilter
|
Output File Names |
Description |
|
CNVR_ALL_ReviewedCNVRs.txt |
50 column significant case
enriched CNVRs with Red Flag annotation for quality filtering |
|
CNVR_ALL_ReviewedCNVRs_brief.txt |
10 column significant case
enriched CNVRs with Red Flag annotation for quality filtering |
|
CNVR_ALL_ReviewedCNVRs_verbose.txt |
100 column significant case
enriched CNVRs with Red Flag annotation for quality filtering (need for TDT
details) |
|
CNVR_ALL_ReviewedCNVRs_ControlEnriched.txt |
50 column significant control
enriched CNVRs with Red Flag annotation for quality filtering |
|
CNVR_ALL_ReviewedCNVRs_brief_ControlEnriched.txt |
10 column significant control
enriched CNVRs with Red Flag annotation for quality filtering |
|
CNV_UCSC_bedfile_ForCustomTrackReview.txt |
.rawcnv significant locus
visualization of individual sample CNVs genome.ucsc.edu Be sure you select
correct genome assembly→click manage custom tracks→Browse for
UCSC bedfile→Paste CNVR coordinates in Genome Browser search box |
|
CNV_UCSC_bedfile_ForCustomTrackReview_Probes.txt |
Probe coverage significant locus
visualization |
|
CNV_ContributingCalls.txt |
CNVRs contributing calls full
boundaries as in the .rawcnv input |
|
CNV_brief.stats |
Probe based CNV association
statistics file |
|
Burden.txt |
Cases and controls IDs, and
TotalDelLengths, and TotalDupLengths |
|
ParseCNV.log |
ParseCNV log |
|
ChrSnp0PosSorted_UseWithPed.map |
Map Sorted to use with CNV ped |
Deletion and Duplication significant CNVRs with
counts in cases and control and p-value. Sample IDs, CN states, CNV calls
contributing to each association and many other statistics and tracking data
are provided (100 fields in total verbose output and 40 highly informative
fields in brief output).
UCSC custom track bed file with CNV calls.
CNV_ContributingCalls.txt: CNV contributing calls for
each CNVR
CNVDEL.ped CNVDUP.ped: .ped files for further
statistics deletion and duplication are generated for use in Plink.
CNVR_ALL_ReviewedCNVRs: All significant CNVRs with CNVType,
redFlagCount, and redFlagReasons.
Nomenclature:
CNVR: CNVRegion intersection region of overlapping CNVs with significant
enrichment in cases vs. controls if overlap (Primary importance, refined CNV
association region)
CNVRange: CNVRange union region of overlapping CNVs
(Typical meaning of “CNVR” in literature)
People are often confused by the small range and
value of the first 2 columns: CNVR and CountSNPs. This is the most significant
overlapping region, not the entire span of contributing CNVs. CNVRange and
AvgNumSnps columns reflect the entire span of contributing CNVs. CNVs at a
particular locus do not have precisely the same boundaries, especially rare
CNVs (see paper Figure 3).
temp/CNV_Verbose.stats full SNP based statistics for
specific locus queries not among the significant loci or for querying
replication based on TagSNP (i.e. GeneName → PositionRange → SNP
IDs in PositionRange → egrep SNP IDs from temp/CNV_Verbose.stats →
CNVFrequencyInGene). A cleaner way is: perl GeneRef/RemoveSpecificBarcodes-Hash_Example_DefFile_ColRemKeep.pl SNPIDsToReplicate.txt temp/CNV_Verbose.stats
1 keep
(FilterCNV.pl)
For querying replication based on genomic coordinate
range use FindSnpsInRange.pl StatsFile Chr Start Stop (much better than grep
cytoband since not specific enough).
AllRes.pdf: Image created with BAF and LRR of
specific association regions across contributing cases with --idToPath option
If you get the error: “-bash: convert: command not
found” run: “sudo apt-get install imagemagick”. This command combines the BAF
LRR plots into a single image to scroll through CNVRs sorted by significance.
If you do not have administrative privileges to do sudo:wget http://www.imagemagick.org/download/ImageMagick.tar.gz
tar
xvzf ImageMagick.tar.gz
cd
ImageMagick-6.8.9-7/
./configure
make
DESTDIR=~/ImageMagick
make install
export PATH=$PATH:~/ImageMagick/usr/local/bin/
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:~/ImageMagick/usr/local/lib/
Alternatively, the images can be flipped through in
Powerpoint using Insert-PhotoAlbum or Open Office Impress Add On
If you have a local R install due to lack of
administrator privileges on your system, you need to provide the full path to
the R executable such as "/work/1a/joe/R/R-3.1.2/bin/R". This path
needs to replace "R" in instances of "R CMD" in these 3
scripts:
ParseCNV.pl
MakeRedFlagPlots.pl
PerlModules/visualize_cnv.pl
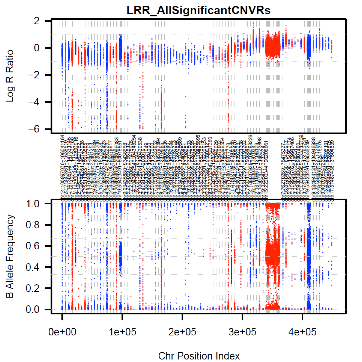
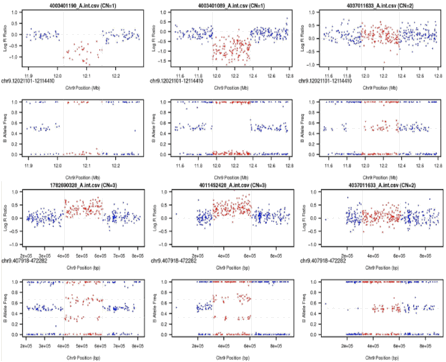
5x10-4 is a conservative bar for CNV
genome-wide significance surviving multiple testing correction based on
analysis of Illumina and Affymetrix genome-wide SNP arrays. The typical bar of
5x10-8 used in GWAS is not appropriate for CNV considering:
§
the
number of probes with a nominal frequency of CNV occurrence (only probes with
some CNV detected are informative)
§
the
number of probes with enrichment in cases vs. controls and vice versa (evidence
of more case enriched loci than control enriched loci)
§
probes
with less than 1% population frequency of CNV (optionally)
§
the
number of CNVRs (multiple probes are needed to detect a single CNV and should
not count separately for multiple testing correction)
2x10-5 according to permutation studies.
UCSC Track Review for spurious association. (UCSCGenomeBrowser-Select
Assembly-add custom tracks-Browse-Submit).
Recommended UCSC tracks:
Header:Mapping and Sequencing Tracks-Item: GC Percent (full) (exclude if low or high across
window)
Header:Variation and Repeats-Item: DGV Struct Var (pack)
and Item:Segmental Dups (pack) (exclude if 10+)
Header:Genes and Gene Prediction Tracks-Item: UCSC
Genes (pack) (See redFlagCount and redFlagReasons columns in “CNVR_ALL_ReviewedCNVRs.txt”
for automated scoring of relevant features originally visually scored).
Header:Phenotype and Literature-Item: ISCA, DECIPHER
BAF/LRR Review of specific association region across
many samples using SpecificBafLrrAccess output AllRes.txt or CNVR specific
file.
BAF/LRR Review of whole chromosome data of sample
with associated CNV and SNP clusters using Illumina Genome Studio Genome Viewer
or Affymetrix Genotyping Console Browser.
qPCR/ddPCR Wet Lab Review for confirmation of true
positive and potentially true negatives.
These are done in order of increasing effort per
locus but the number of loci will be filtered down by each step.
Red flag count <= 3 may be considered high
confidence results. More important red flags to fail a CNVR as low confidence
include: AvgProbes<5, DgvEntries>10, PenMaxP>0.5 and high frequency,
SegDups>10, Recurrent, FreqInflated,
AvgConf<10, ABFreqLow.
Standard Filter for high significant and confident
results for SpecificBafLrr Visual: Sort RF <=3, delP<5x10^-4 and
ORDup>1 OR dupP<5x10^-4 and ORDup>1, (on exon)
|
RedFlag |
Default Report
Threshold |
Explanation |
|
SegDups (count, max, avg) |
>10, >0.98 max Fraction Matching |
Many segmental duplications (i.e., nearly
identical DNA segments), representing genomic segments that are difficult to
uniquely hybridize probes to, which could underlie false positive CNV
detection. Segmental
Duplications inform CNV breakpoints if flanking (include) and noisy regions
if overlapping (exclude). |
|
DgvEntries |
>10 |
Overlapping multiple Database of Genomic
Variants (DGV) entries, representing CNV signals observed in “healthy”
individuals, suggesting that a potential association result in the study at
hand may be false. |
|
TeloCentro |
any overlap |
Residing at centromere and telomere proximal
regions as they often have sparse probe coverage and only have a single
flanking diploid reference to base CNV calls. |
|
AvgGC |
31>GC>60 |
Harboring high or low GC content regions that
bias probe hybridization kinetics even after GC model correction is done by
CNV calling algorithms, producing false CNV calling and biasing the result. |
|
AvgProbes |
<10 |
CNVs captured with low average number of
probes, contributing to association with low confidence. If an association
depends on a preponderance of small CNVs, the likelihood of false positive is
high. |
|
Recurrent |
any overlap |
Locus frequently found in multiple studies
such as TCR, Ig, HLA, and OR genes. TCRs undergo somatic rearrangement due to
VDJ recombination causing inter-individual differences in the clonality of
T-cell populations and thus are not true CNVs, necessitating exclusion. |
|
PopFreq |
>0.01 |
CNV regions with high population frequency
(for rare CNV focused studies) indicate that probe clustering is likely
biased due to a high percentage of samples with CNV used in clustering
definition thus biasing CNV detection. |
|
PenMaxP_Freq_HighFreq |
PenMaxP >0.5, Freq >0.5, HighFreq >0.05 |
CNV peninsula of common CNV (sparse probe
coverage and nearby high frequency CNV) indicates that within the range of
contributing CNV boundaries there is a non-significant (p>0.5) p-value
which is notably different from the CNVR association typically due to random
extension of common CNVs to neighboring sparse or noisy probes. PenMaxP is the worst
p-value in the span of CNV calls contributing to the significant CNVR. Freq
is the frequency of this PenMaxP worst p-value. HighFreq is the frequency any
non-nominally significant p-value (P>0.05). |
|
FreqInflated |
>0.5 sids at this locus have
>(maxInflatedSampleCount-2) occurrences in all significant results |
The same inflated sample driving multiple CNV
association signals. Certain samples have many noisy CNV calls arising in
rare regions despite upfront sample quality filtering. |
|
Sparse |
>50kb |
A large gap in probe coverage exists within
the CNV calls indicating uncertainty in the continuity of a single CNV event,
typically due to dense clusters of copy number (intensity only) probes with
large intervening gaps. |
|
ABFreq |
<1% values (0.1,0.4) or (0.6,0.9) |
For duplications, AB banding of BAF at 0.33
and 0.66 for CN=3 or 0.25 and 0.75 for CN=4 are very important observations
given the relatively modest gain in intensity observed in duplications. |
|
AvgConf |
<10 |
The HMM confidence score in PennCNV is a
superior indication of CNV call confidence compared to numsnps and length in
studies comparing de novo vs. inherited CNV calls, giving an indication of
the strength of the CNV signal or aggregate difference in probability between
the called CN and the next highest probability CN. Other CNV calling
algorithms give different range confidence scores or lower values might mean
more confidence (i.e. call p value) so threshold may need modification. It is
recommended to be in .rawcnv file as column 8 i.e. “conf=20.659” but not
required. |
|
AvgLength |
<10kb |
A classical confidence scoring parameter is
the length of the CNV. If the CNV is too small, it is submicroscopic and even
if many probes are tightly clustered, bias of local DNA regions and probe
overlap make confidence difficult. |
If you did the case:control (default), you want to
look at “DelTwoTail” and “DupTwoTail”.
If you did the --tdt, you want to look at “TDTDel”
and “TDTDup” for TDT P-value and “NormParDel” and “NormParDup” for the count de novo.

Sequencing

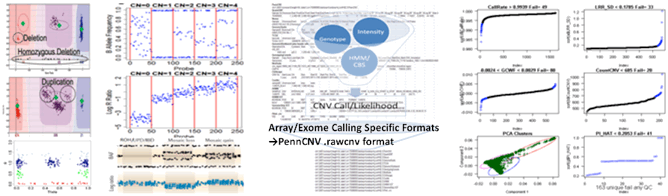
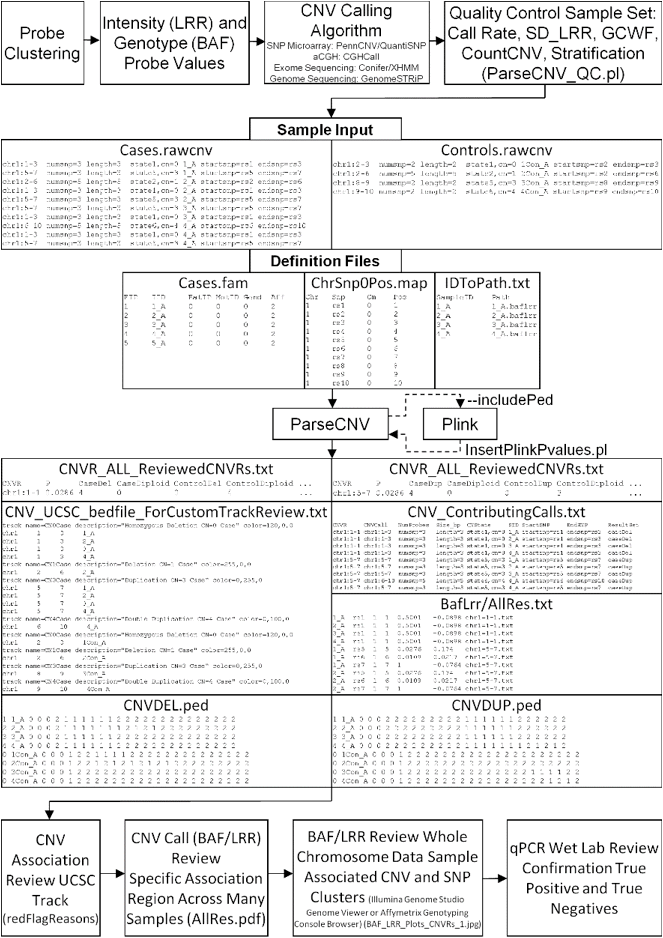
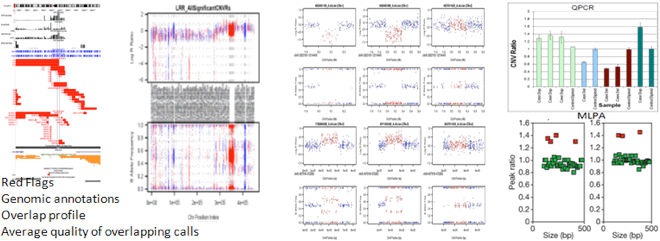
Significant CNVR
Output Fields Description:
|
Column |
Description |
|
CNVR |
CNV Region of greatest significance and
overlap coordinates |
|
CountSNPs |
The number of probes available in the CNVR for
this dataset In this case, contributing individual CNV calls may be larger |
|
SNP |
Tag SNP for ease and clarity of reporting and
replication |
|
DelTwoTailed |
Two Tailed Fisher's Exact P-value based on the
contingency table Cases Del/Cases Diploid/Controls Del/Controls Diploid as
listed separately |
|
DupTwoTailed |
Two Tailed Fisher's Exact P-value based on the
contingency table Cases Dup/Cases Diploid/Controls Dup/Controls Diploid as
listed separately |
|
ORDel |
The Odds Ratio for deletion. |
|
ORDup |
The Odds Ratio for duplication. |
|
Cases Del |
The number of cases with a deletion detected
in this region by PennCNV |
|
Cases Diploid |
The number of cases without a deletion or
duplication detected in this region by PennCNV |
|
Control Del |
The number of controls with a deletion
detected in this region by PennCNV |
|
Control Diploid |
The number of controls without a deletion or
duplication detected in this region by PennCNV |
|
Cases Dup |
The number of cases with a duplication
detected in this region by PennCNV |
|
Cases Diploid |
The number of cases without a deletion or
duplication detected in this region by PennCNV |
|
Control Dup |
The number of controls with a duplication
detected in this region by PennCNV |
|
Control Diploid |
The number of controls without a deletion or
duplication detected in this region by PennCNV |
|
IDsCasesDel |
The sample IDs of cases corresponding to the
Cases Del column for clinical data lookup. To convert to list in Excel:
Data-TextToColumns-Delimited-Space then Copy-PasteSpecial-Transpose |
|
IDsCasesDup |
The sample IDs of cases corresponding to the
Cases Dup column for clinical data lookup. To convert to list in Excel:
Data-TextToColumns-Delimited-Space then Copy-PasteSpecial-Transpose |
|
StatesCasesDel |
CN states listed corresponding to IDsCasesDel
(1(CN=0)/2(CN=1)) |
|
StatesCasesDup |
CN states listed corresponding to IDsCasesDup
(5(CN=3)/6(CN=4)) |
|
TotalStatesCases(1) |
The number of cases in Cases Del with a
homozygous deletion or both copies lost |
|
TotalStatesCases(2) |
The number of cases in Cases Del with a
hemizygous deletion or one copy lost |
|
TotalStatesCases(5) |
The number of cases in Cases Dup with a
hemizygous duplication or one copy gained |
|
TotalStatesCases(6) |
The number of cases in Cases Dup with a
homozygous duplication or two copies gained |
|
IDsDelControl |
The sample IDs of controls corresponding to
the Control Del column for clinical data lookup. |
|
IDsDupControl |
The sample IDs of controls corresponding to
the Control Dup column for clinical data lookup. |
|
StatesDelControl |
CN states listed corresponding to
IDsDelControl (1(CN=0)/2(CN=1)) |
|
StatesDupControl |
CN states listed corresponding to
IDsDupControl (5(CN=3)/6(CN=4)) |
|
TotalStates(1) |
The number of Controls in Controls Del with a
homozygous deletion or both copies lost |
|
TotalStates(2) |
The number of Controls in Controls Del with a
hemizygous deletion or one copy lost |
|
TotalStates(5) |
The number of Controls in Controls Dup with a
hemizygous duplication or one copy gained |
|
TotalStates(6) |
The number of Controls in Controls Dup with a
homozygous duplication or two copies gained |
|
ALLTwoTailed |
All CNV states considered together p |
|
ORALL |
All CNV states considered together OR |
|
ZeroTwoTailed |
Only CN=0 CNV state considered together p |
|
ORZero |
Only CN=0 CNV state considered together OR |
|
OneTwoTailed |
Only CN=1 CNV state considered together p |
|
OROne |
Only CN=1 CNV state considered together OR |
|
ThreeTwoTailed |
Only CN=3 CNV state considered together p |
|
ORThree |
Only CN=3 CNV state considered together OR |
|
FourTwoTailed |
Only CN=4 CNV state considered together p |
|
ORFour |
Only CN=4 CNV state considered together OR |
|
Gene |
The closest proximal gene based on UCSC Genes
which includes both RefSeq Genes and Hypothetical Gene transcripts |
|
Distance |
The distance from the CNVR to the closest
proximal gene annotated. If the value is 0, the CNVR resides directly on the
gene. |
|
Description |
The gene description delimited by
"/" for multiple gene transcripts or multiple genes listed |
|
Pathway |
Annotated pathway membership of Gene with
reference compiled from Gene Ontology database, BioCarta database and the
KEGG database (definition files in GeneRef folder) |
|
AverageNumsnpsCaseDel |
The average numsnp of CNV calls contributing
to Case Del CNVR. Allows for much more informative CNV size (confidence)
filtering post-hoc. |
|
AverageLengthCaseDel |
The average length of CNV calls contributing
to Case Del CNVR. Allows for much more informative CNV size (confidence)
filtering post-hoc. |
|
CNVRangeCaseDel |
Alternative larger CNV Range Case Del definition compared to
minimal common overlap definition of CNVR |
|
AverageNumsnpsControlDel |
The average numsnp of CNV calls contributing
to Control Del CNVR. Allows for much more informative CNV size (confidence)
filtering post-hoc. |
|
AverageLengthControlDel |
The average length of CNV calls contributing
to Control Del CNVR. Allows for much more informative CNV size (confidence)
filtering post-hoc. |
|
CNVRangeControlDel |
Alternative larger CNV Range Control Del definition compared to
minimal common overlap definition of CNVR |
|
CNVType |
Deletion or duplication CNVR Significant in
combined report |
|
Cytoband |
Cytoband genomic landmark designations |
|
redFlagCount |
Count red flag from association review of 9
(see text, briefly: SegDups, DGV, Centro/Telo, GC, ProbeCount, PopFreq, Peninsula,
Inflated) |
|
redFlagReasons |
The failing metrics for association review and
their values |
Reformat CNV Calls
from Different Algorithms/Sources into Standard PennCNV .rawcnv Format:
CNV Calling Algorithms Formats:
PennCNV
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 PennCNV
QuantiSNP
Sample Name Chromosome Start
Position (bp) End
Position (bp) Start
Probe ID End
Probe ID Length
(bp) No.
Probes Copy
Number Max.
Log BF Log
BF: State 0 Log
BF: State 1 Log
BF: State 2 Log
BF: State 3 Log
BF: State 4 Log
BF: State 5 Log
BF: State 6
7500809055_R02C01 1 248740572 248795110 kgp8531759 kgp6234332 54539 19 0 137.858 137.858 -9.05444 0 1.18944 -7.74257 -9.26705 -44.1485
Nexus
Sample Chromosome Region Event Length Cytoband % of CNV Overlap Probe Median %
Heterozygous Probes Min Size Min Region Max Size Max Region Locus
IDs Call Pvalue
4363511659_R02C01 chr3:191,219,380-191,224,532 Homozygous Copy Loss 5153 q28 84.14208075 -4.471399784 40 5
GenomeStudio
SampleID BookmarkType Chr Start End Size Author CreatedDate Value Comment
K069 [735] CNV
Bin: Min 0 To Max 0.5 1 17548878 17549783 905 2011-07-13
오후 2:15:11 0 CNV
Confidence: 177.519
TCGA
barcode chromosome start stop num.mark seg.mean
TCGA-13-0891-01A-01D-0403-02 Y 22008576 22014412 -11.7734
GenomicWorkbench (also header with colons and footer with equals)
AberrationNo Chr Cytoband Start Stop #Probes Amplification Deletion pval Gene
Names
AS_001
1620 chr5 q23.3 127363880 127364126 3 0 -5.862602 0
GenotypingConsole
Sample Copy Number State Loss/Gain Chr Cytoband_Start_Pos Cytoband_End_Pos Size(kb) #Markers Avg_DistBetweenMarkers(kb) %CNV_Overlap Start_Linear_Pos End_Linear_Position Start_Marker End_Marker CNV_Annotation
Genomewide6.0_120.CN5.CNCHP 0 Loss 4 q13.2 q13.2 112 61 2 100 69371991 69484097 CN_1109547 CN_1111668 Variation_1082
// chr4:69428222-69459530 // Mendelian inconsistencies //
BirdSuite
cnv_region_id 10893_INNED_g_Plate_No._6_GenomeWideSNP_6_E07_52176.CEL 11109_PERDU_g_Plate_No._30_GenomeWideSNP_6_G04_51146.CEL 11213_ARDOR_g_Plate_No._31_GenomeWideSNP_6_G10_51844.CEL 11888_JOYED_g_Plate_No._7_GenomeWideSNP_6_C10_46602.CEL 12233_CRAVE_g_Plate_No._25_GenomeWideSNP_6_F12_50312.CEL 12643_STAYS_g_Plate_No._1_GenomeWideSNP_6_H02_54228.CEL 12719_CRAVE_g_Plate_No._25_GenomeWideSNP_6_G02_50154.CEL 12864_SCHWA_g_Plate_No._3_GenomeWideSNP_6_F09_54144.CEL 13534_ARDOR_g_Plate_No._31_GenomeWideSNP_6_F12_51874.CEL #chrom chromStart chromEnd region
CNP10 2 2 2 2 2 2 2 2 2 chr1 755964 799636 CNP3
CGHscape
CNV No Gain/Loss Chr Size Start
ID Start Pos End ID End Pos Length Score
1 Loss chr18 21011675 RP11-178F10 20300971 RP11-463D17 41312645 21 1.03776
Conifer
sampleID chromosome start stop state
1-00079_BEST.sorted.bam.rpkm chr1 104205278 104238261 dup
XHMM
SAMPLE CNV INTERVAL KB CHR MID_BP TARGETS NUM_TARG Q_EXACT Q_SOME Q_NON_DIPLOID Q_START Q_STOP MEAN_RD MEAN_ORIG_RD
1-00079 DEL chr3:100566432-100570801 4.37 chr3 100568616 39697..39701 5 24 36 36 17 20 -3.41 254.73
XHMM VCF
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 1-00070
#chr1 1386051 chr1:1386051-1431197 <DIP> <DEL>,<DUP> . . AC=11,1;AF=0.04,0.00;AN=279;END=1431197;IMPRECISE;SVLEN=45147;SVTYPE=CNV;TPOS=1386051;TEND=1431197;NUMT=27;GQT=4 GT:NDQ:DQ:EQ:SQ:NQ:LQ:RQ:PL:RD:ORD:DSCVR 0:0:69:0,0:0,0:95,69:0,0:0,0:0,255,255:0.65:62.72:N
GenomeStrip VCF
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT HG00096 HG00100 HG00103 HG00106 HG00108
1 738636 MERGED_DEL_2_47 A <DEL> . . CIEND=-17,33;CIPOS=-18,19;END=742073;SOURCE=GenomeStrip_738636_742073;SVTYPE=DEL GT:FT:GL:GQ 0/0:LowQual:-0.11,-0.65,-17.33:5.35 0/1:LowQual:-0.38,-0.24,-5.92:1.38 0/0:LowQual:-0.05,-0.94,-22.07:8.92 0/1:LowQual:-0.63,-0.12,-3.49:5.09 0/0:PASS:-0.00,-2.32,-36.68:23.15
Plink .cnv
FID IID CHR BP1 BP2 TYPE SCORE SITES
1 AU000103 10 46410734 47138713 4 5 24
CNVnator / SG-ADVISER (Does not specify sample ID)
Chromosome Begin End VarType Notes
chr1 45303803 45307152 loss -
chr1 93256031 93308213 gain -
Generic Feature Format 3 http://gmod.org/wiki/GFF3
Seqid source type start end score strand phase attributes
##gff-version 3
##gvf-version 1.07
chr1 PennCNV CN0
1300 1500 82.372 +
. .
dnaCopy
Chr Start Stop Num_Probes Segment_Mean
1 1300 1500 20
-4
Affymetrix ChAS
File CN
State Type Chromosome Min Max Size
(kbp) Mean
Marker Distance Marker
Count Max
% Overlap Overlap
Map Items (% of Segment overlapped) CytoRegions Use
In Report Interpretation Confidence Cytoband
Start Cytoband
End Genes DGV FISH
Clones sno/miRNA BACs OMIM
® Segmental Duplications Smoothed/Joined
A.cyhd.cychp 3 Gain 5 76152823 76341069 188.246 1140 166 . Overlap
Map Not Set Cytoregions
Not Set TRUE . 0.8990996 q13.3 q13.3 S100Z,
CRHBP, AGGF1 Variation_47921,
Variation_6416, Variation_111241, Variation_64234 RP11-206N2,
RP11-62D9 . RP11-15O12,
RP11-600J9, RP11-1129G24, RP11-1001I23, CTC-775M8, RP11-30D22, RP11-996M9,
CTA-344D19, CTD-2065N18, CTD-2379B23, CTD-2378O19, CTD-3165C22, CTD-3027I16,
RP11-372K5, RP11-139O19, CTD-3169C4, CTD-2100D3, CTD-2294O13, RP11-62D9,
RP11-372H22, CTD-2219B18, RP11-701A4, RP11-158F8, RP11-432J21, CTD-2169P21,
CTD-3215E19, RP11-771N6, CTD-3241E15, RP11-884J13 . . FALSE
1000 Genomes format (Critical: CHR,START_INNER,END_INNER,TYPE_OF_EVENT,
SIZE_PREDICTION, and SAMPLES)
CHR START_OUTER START_INNER END_INNER
END_OUTER
TYPE_OF_EVENT
SIZE_PREDICTION MAPPING_ALGORITHM
SEQUENCING_TECHNOLOGY
SAMPLEs TYPE_OF_COMPUTATIONAL_APPROACH GROUP OPTIONAL_ID
1
829757 829757 829865 829865 DEL 116 MAQ SLX NA19238,NA19240 RP WashU
BreakDancer format
#Chr1 Pos1 Orientation1 Chr2 Pos2 Orientation2 Type Size Score num_Reads num_Reads_lib example1.bam example2.bam
10 89690279
+
10
89702321
+
DEL 12042 99 16 example1|16 0.01 2.37
10 85512695
+
10
85513886
+
DEL
1191 99 18
example1|11:example2|7
0.02 0.35
Bedpe
Chr1 Start1 End1 Chr2
Start2 End2 Name Score Strand1 Strand2
chr1 100 200 chr5 5000 5100 bedpe_example1 30 + -
chr9 1000 5000 chr9 3000 3800 bedpe_example2 100
+ -
Reformatted to PennCNV .rawcnv Example:
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 PennCNV
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 QuantiSNP
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 Nexus
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 GenomeStudio
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 TCGA
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 GenomicWorkbench
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 GenotypingConsole
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 BirdSuite
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 CGHscape
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 Conifer
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 XHMM
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 XHMM
VCF
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 GenomeStrip
VCF
chr1:248740572-248795110 numsnp=20 length=54,539 state1,cn=0 7500809055_R02C01 startsnp=kgp8531759 endsnp=kgp6234332 conf=82.372 Plink
.cnv
If you really would like to see a specific feature or
have an idea how to optimize please email me.
De novo Note: On 2012-09-12 I introduced TDT p-value
driven CNVR definition to use alternatively to Case-Control. The third popular
route of study is de novo but there is no de novo p-value, simply a count which
is rarely real upon review at 2 or more cases on one locus, making de novo or
large CNV Burden and Gene Set Enrichment Analysis by DAVID, DAPPLE, or
Ingenuity more popular and fruitful lines of investigation. However, I feel
these are not robust alternatives. Currently, see the --tdt CNVR result columns
“NormParDel” and “NormParDup” for the count de novo (Now DenovoDel and
DenovoDup). A true de novo p-value would be minor allele deletion/duplication
(de novo + transmitted + cases incomplete trio) vs. (untransmitted + controls).
Do you prefer probabilistic CNV genotyping as in
CNVTools rather than discrete as in PennCNV? Maybe you could run PennCNV
-validate to get likelihoods and I could output a SNPTEST .geno file and
InsertSnpTestPvalues to mark up red flags. perl detect_cnv.pl -validate -hmm
example.hmm -pfb example.pfb -log ex5.log -out ex5.rawcnv -candlist ccnvr -list
inputlist -valilog ex.valilog
ArrayConcordance_InheritanceDenovo_AnnotateCNVCalls/PQN_Concord_Inh.pl:
Generate
new overlapping segments consensus CNV calls
from multiple algorithm comparison for further analysis.
SKAT test statistic to increase sensitivity http://cran.r-project.org/web/packages/SKAT/
PathwayBased/GeneBased to increase sensitivity of
multi-genic non-overlapping signals with RedFlags and ped output for
specificity and stratification correction
Continuous RedFlag to increase specificity robustly
correlating to validation and true association? Pass/Fail annotation based on RedFlags?
ParseCNV --includeAllRedFlags
plot R histogram: using MakeRedFlagPlots.pl CNVR_ALL_ReviewedCNVRs_brief.txt
plot R curve: using lines(density(a$a))
integrate observed value at significant CNVR
in proper direction of red flag: dens2 <- density(a$SegDups, from=0, to=a$SegDups[i]) with(dens2, sum(y * diff(x)[1]))
average integration likelihoods: Not very
well correlated with validation success by simple average (same weights).
Correlate/Weight with validation success: GLM
weights assigned and correlation of 0.8 with validation success achieved with
reasonable cutoff for GLMWeightedConfidence of 0.2. ROC curve looks
solid and the AUC score is 0.983 using ROCR.
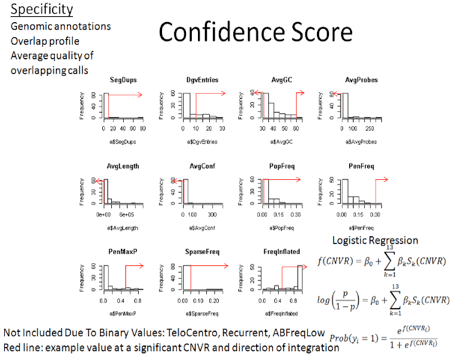

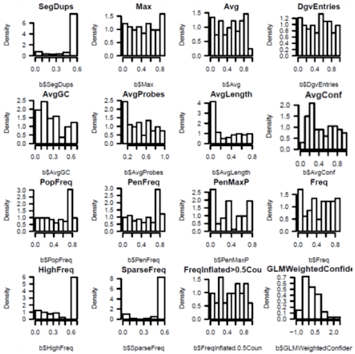

Combined UCSC, BAF LRR, SNP Clusters and PCA with CNV
samples highlighted for passing RedFlag Confidence and Multiple Testing
Correction Significance
Integrate TADA: Transmission And De novo Association
Test
Epept permutation p-value for genome wide significance
adjustment and fast low p-values
Permutation
p-values typically take a long time to calculate, do not differentiate very
significant p-values
FaST-LMM-Select – Rare variant
stratification correction
ParseCNV uses PercentSamples.pl as a
module for –batch
Report multiple CNVRs within 1MB if
multiple pass multiple testing correction threshold.
CNV Workshop, CNVannotator,
CNVAnalysisToolkit
SG-ADVISER: The annotation results
include details regarding location, impact on the coding portion of genes,
allele frequency information (including allele frequencies from the Scripps
Wellderly cohort), and overlap information with other reference data sets
(including ClinVar, DGV, DECIPHER). A summary variant classification is
produced (ADVISER score) based on the American College of Medical Genetics and
Genomics scoring guidelines. We demonstrate >90% sensitivity/specificity for
detection of pathogenic events.
http://www.geneimprint.com/site/genes-by-species CNVs create problems especially for imprinted loci which only express the mother or father copy.
CNV disease database (Similar to NHGRI GWAS Catalog but for CNVs)
http://bioinfo.mc.vanderbilt.edu/CNVannotator/database/CNVD/cnvd_hsa.good2db.gene
http://202.97.205.78/CNVD/download.jsp
http://bedtools.readthedocs.org/en/latest/content/general-usage.html
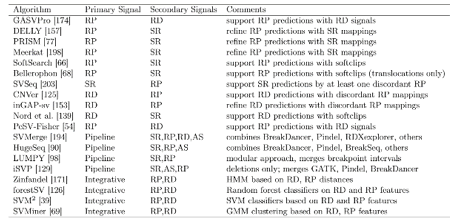
Mosaicism calls from R-GADA or BAFSegmentation.
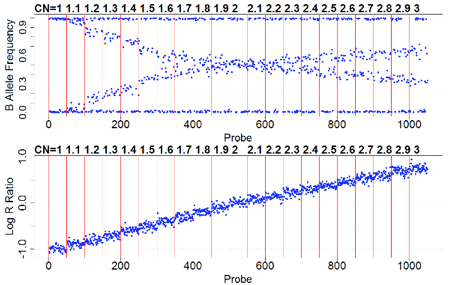
Replace arrays with pointers, binary, or indexed
memory/files for less RAM and disk space (ped vs. bed) footprint. Less RAM
would allow genome wide runs using any array resolution and cohort size without
by chromosome splitting. Advice on that would be much appreciated.
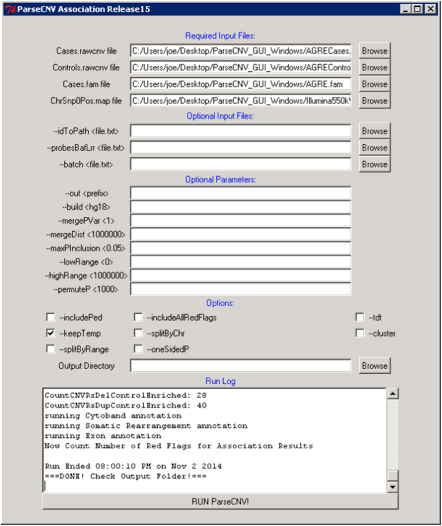
Wx Perl or Tk Perl Needed:
perl -MCPAN -e “install Wx” I had some trouble installing Wx on
a machine but it is more portable and uses native GUI elements of the system
when made into .exe.
perl -MCPAN -e "install
Tk" Tk
is more established and has more documentation than Wx.
Cygwin “couldn’t connect to
display” error: Install the cygwin packages “xorg-server” and “xinit” and type
“startxwin” to start an X terminal and “perl ParseCNV_GUI_Tk.pl” will launch
the window.
Idea: Web server to host the
tool in a website.
Updated CNVRuler Supplementary Table 1. Comparison of the CNV-association analysis
Tools
|
|
CONAN |
BirdSuite |
Plink |
CNVineta |
CNVassoc |
CNVTools |
R-Gada |
CNVRuler |
HD-CNV |
ParseCNV |
|
Input Platform |
Affymetrix |
Affymetrix |
ALL |
Illumina Affymetrix |
ALL |
ALL |
ALL |
ALL |
ALL |
ALL |
|
CNV Call Data |
PennCNV |
Array data |
PED1) |
APT1) |
CGHcall |
Text file1) |
BeadStudio1) |
Nexus |
CSV |
Nexus |
|
|
QuantiSNP |
|
BirdSuite2) |
QuantiSNP1) |
Plink |
|
Genotyping Console1) |
PennCNV |
|
PennCNV |
|
|
Genotyping Console |
|
|
|
Text file1) |
|
Text file1) |
Genomic Workbench |
|
Genomic Workbench |
|
|
Text file1) |
|
|
|
|
|
|
TCGA |
|
TCGA |
|
|
MS Exel1) |
|
|
|
|
|
|
NimbleScan |
|
NimbleScan |
|
|
|
|
|
|
|
|
|
APT |
|
APT |
|
|
|
|
|
|
|
|
|
Genotyping Console |
|
Genotyping Console |
|
|
|
|
|
|
|
|
|
Genome Studio |
|
Genome Studio |
|
|
|
|
|
|
|
|
|
Text file |
|
Text file |
|
OS |
ALL |
Linux |
ALL |
ALL |
ALL |
ALL |
ALL |
ALL |
ALL |
ALL |
|
Frequent CNV Region3) |
CNVR |
N/A |
N/A |
Fragment |
CGHregions |
N/A |
N/A |
CNVR |
CNVR |
CNVR |
|
|
|
|
|
|
|
|
RO |
|
RO |
|
|
|
|
|
|
|
|
|
|
Fragment |
|
Fragment |
|
GUI |
Yes |
No |
Yes4) |
No |
No |
No |
No |
Yes |
Yes |
Yes |
|
Required |
Oracle (Optional) |
Matlab |
No |
R |
R |
R |
R |
R |
Java Swing |
R |
|
|
Annotation File |
R |
|
|
|
|
|
|
JGraphT |
|
|
|
|
Annotation File |
|
|
|
|
|
|
|
|
|
Statistical Methods |
Linear regression |
Regression |
CA Trend Test |
Logistic regression |
Logistic regression |
Maximum likelihood |
Logistic regression |
Fisher’s exact test |
Interval Graph |
Fisher’s exact test |
|
|
|
(SNP ref) |
Fisher’s exact test |
|
Linear regression |
EM |
Likelihood Ratio Test |
Chi-Square |
Bron Kerbosch Clique Finder Algorithm |
Chi-Square |
|
|
|
|
Stratified Test |
|
|
|
|
Logistic regression |
Gephi |
CA Trend Test |
|
|
|
|
Multi-locus Test |
|
|
|
|
Linear regression |
|
Stratified Test |
|
|
|
|
Likelihood Ratio Test |
|
|
|
|
|
|
Multi-locus Test |
|
|
|
|
Logistic regression |
|
|
|
|
|
|
Likelihood Ratio Test |
|
|
|
|
Linear regression |
|
|
|
|
|
|
Logistic regression |
|
|
|
|
|
|
|
|
|
|
|
Linear regression Confidence Score |
|
Disadvantage / Limitation |
Support Platform |
Support Platform |
Data conversion |
Support Platform |
Data conversion |
Data conversion |
User Interface |
Graphical Report |
P-value |
Graphical Report |
|
|
Single Statistical Method |
Large data handling |
Region definition |
Single Statistical Method |
User Interface |
Region definition |
|
|
Confidence Scoring |
|
|
|
|
Region definition |
|
User Interface |
|
User Interface |
|
|
|
|
|
|
|
User Interface |
|
|
|
No Covariates |
|
|
|
|
|
|
|
|
|
|
|
Limited data model |
|
|
|
|
|
|
|
|
|
|
|
(Binary, |
|
|
|
|
|
|
|
|
|
|
|
Normal distribution) |
|
|
|
|
|
Reference |
Forer et al, 2010 |
Korn et al, 2008 |
Purcell et al, 2007 |
Wittig et al, 2010 |
Subirana et al, 2011 |
Barnes et al, 2008 |
Pique-Regi et al, 2010 |
Kim et al, 2012 |
Butler et al, 2012 |
Glessner et al, 2013 |
|
1) Manual
Conversion required |
||||||||||
|
2) Supported by
BirdSuite |
||||||||||
|
3) Since each
tool named differently for identical region definition, the representative
words are chosen from this study for convenience |
||||||||||
|
4) Supported by
3rd party front-end gPlink |
||||||||||
2019-07-01: ParseCNVRelease_21
Posted: ABFreq Fixed to be 0-1
as expected in ParseCNV.pl lines 5985-5988
where $#Indexes+1 is
number of snps and $i-1 is number of samples at this CNVR.
Include ./GeneRef/hg19_cytoBand_SimFormat_AllCol.sorted
OutputUtilities/PercentSamples.pl
space before and after countcohortsid to allow for separate counting of
categories which are substrings of other categories
2018-01-04: ParseCNVRelease_20
Posted: Mace et al quality
score calculation integrated into ParseCNV.pl when PennCNV --log specified.
ParseCNV_QC now does
CNV Call QC in addition to Sample QC. : numsnps length conf mace
CNV impact CADD with
SVScore
Annotation from clinical
CNV databases including Decipher and ClinGen.
2016-08-17: ParseCNVRelease_19
Posted: Improved Folder Design
for Clarity of Structure and Role of files and scripts
ParseCNV_QC cleaned up
Cases.rawcnv only
specify option for simplicity
2015-08-23: ParseCNVRelease_18
Posted: .bim creation converts
X->23, Y->24, XY->25, M/MT->26
--sub to allow qsub or
bsub depending on the type of cluster you have and –clusterHeader file
specify
--splitByChr now
includes X and Y
MakeRedFlagPlots.pl:
Save Del FreqInflated sample count since Del and Dup CNVRs may have different
count sample cutoffs for FreqInflated
Rm –r temp clean
up to avoid error: cannot remove `temp/<out>': Is a directory
Try to run plink
--make-bed on the individual major bed to make a snp major bed more compatible
with plink
If 0 significant snps,
do not create CNVR
Handling unrecognized
chromosomes in scan_region NOT_FOUND output
speed up the
step: "Make PDF image of BAF/LRR for significant association regions
across all contributing case samples."
HWE p-value
added to Brief.stats
Lessen/Dynamically
define default dependencies in InsertPlinkPvalues.pl
Ensure 100%
progress, list zero controls in the case of NullControl
Include hg38
GeneRef Files
2015-05-14: ParseCNVRelease_17 Posted: --includePed now writes directly to a Plink .bed file to save disk space
--noweb option added
for systems with LWP error
Since only for Dups,
don't include ABFreqLow_High in allredflags which requires a square matrix for
MakeRedFlagPlots
MakeRedFlagPlots.pl
FreqInflated variable name dynamically defined
CNVRangeCaseDel/Dup
header changed to CNVRangeCaseDel/Dup_Indexes
ConvertDGVToRawcnv.pl
ConvertPlinkToRawcnv.pl
ConvertVCFToPennCNV_GenomeStrip_2.pl
http://www.broadinstitute.org/~handsake/mcnv_data/bulk/1000G_phase1_cnv_genotypes_phased_25Jul2014.genotypes.vcf.gz
ConvertVCFToPennCNV_GenomeStrip_3.pl
http://www.broadinstitute.org/~handsake/mcnv_data/bulk/1000G_phase1_cnv_genotypes_unphased_25Jul2014.genotypes.vcf.gz
ParseCNV_GeneBased.pl
includes .tped output
2014-11-03: ParseCNVRelease_16
Posted: Burden.txt includes
CountDel and CountDup per sample
ParseCNV.pl option
--oneSidedP to calculate one sided P assuming only enriched in case CNVs are of
interest
ParseCNV.pl option --permuteP
to permute P number of permutations (takes longer time computationally)
ParseCNV_QC.pl CountCNV
quality threshold bug Fix
Allow id and path to be different by putting the path from idToPath into the .rawcnv file being used for visualize_cnv.pl.
maxDelInflatedSampleCount
bug fix so not reinitialized to zero
detect if UCSC sorting
failed, if so print error message and try again
automatically make <CNVR_ALL_ReviewedCNVRs_brief.txt
ParseCNV --includeAllRedFlags output> input and run perl MakeRedFlagPlots.pl
automatically PassFail
annotation based on Fail any: AvgProbes<5, DgvEntries>10, PenMaxP>0.05
and >0.5 frequency, SegDups>10 or Avg SegDup Match >0.98, any Recurrent,
>0.5 FreqInflated, AvgConf<10, and ABFreqLow <0.01.
calculate Average
Inter Probe Distance to inform --mergeDist setting
GUI_Tk added “Tk” to “my
$mw = Tk::MainWindow->new;” to allow launching window without “couldn’t
connect to display” error.
ParseCNV.pl temp
folder path generated to match input Cases.rawcnv and Controls.rawcnv paths for
sort UCSC
ParseCNV.pl Command
line options maintained in --splitByChr and –splitByRange
ParseCNV_GUI_Windows: PAR::Packer, Inf->9**9**9, DBM::Deep, Editbin /largeadressaware perl.exe, Undef @$arrayID
2014-07-11: ParseCNVRelease_15
Posted: wgetUcscPdfs.sh
<chr start stop id file> <hgsid> added to pull UCSC .pdf images in
batch from CNVRs or CNVRanges as query.
./wgetUcscPdfs.sh
EM_IS_wgetUcscSigPdfs_WD.txt 377963093_ccAa6nbzNe6XDuIygEO9nFJZyevt
gs -q
-sPAPERSIZE=letter -dNOPAUSE -dBATCH -sDEVICE=pdfwrite
-sOutputFile=EM_IS_WD_Significant_CNVRanges.pdf EM_IS_WD*
PercentSamples.pl for
showing contributing counts of batches in IDsCasesDel column.
Automatically make-bed
and delete ped with --includePed to save disk space, Delete Temp Files (except
Verbose.stats) unless --keepTemp used
Burden.txt output with
cases and controls IDs, and TotalDelLengths, and TotalDupLengths
--splitByChr to Split
run by chromosome for large projects that run out of memory
--cluster option
requires a parallel computing cluster you can qsub to (speeds up --splitByChr and
--splitByRange run)
--splitByRange divides
numsnps ranges equally into 22 parts or by density to balance memory
requirements and calls --lowRange and --highRange low and high index for range
specific run if only interested in specific locus or for large memory intensive
projects
%ChrPosToSNP deleted
and %SnpsFoundInOrigMap cleared after usage to save memory
PQN_Concord_Inh.pl checks IDs are present in each algorithm
input and deletes any which are not to avoid shifted comparisons, cluster_header.txt
command line argument to specify file with cluster header specific to
environment, Temp folder with intermediate files
2014-02-24:
ParseCNVRelease_14 Posted: ParseCNV.pl MaxSegdup
and AvgSegdup > 0.98 reporting in addition to count, CNVType [Deletion Duplication
DeletionControlEnriched DuplicationControlEnriched] Not Always included
annotation columns bug fixed with close PEDFILE, InsertPlinkPvalues.pl now allows --build
susScr3 and --build bosTau7, GUI: Paste as separate command instead
of | to prevent GUI Cygwin error, OR
in Brief.stats, ArrayConcordance_InheritanceDenovo_AnnotateCNVCalls/PQN_Concord_Inh.pl
Posted for multiple CNV detection algorithm comparison for filtering of calls
for confidence up front before CNV association testing and de novo,
transmitted, and untransmitted CNV call based annotation to complement CNVR
based annotations, MakeRedFlagPlots.pl: derive continuous confidence score
based on redFlagReasons column in ParseCNV --includeAllRedFlags.
2013-11-22: ParseCNVRelease_13 Posted: Check CN state entries and alert non (0,1,3,4) values, ParseCNV_QC supports general format “SampleID CallRate” GetCallRate.pl AND Run once with no thresholds specified and review automatically determined thresholds based on outliers in your dataset. Then you may specify some adjusted threshold for a given quality metric based on your review of the plots. For example add: --qccallrate 0.98 --qclrrsd 0.2 --qcgcwf 0.005 –qcnumcnv 100 --qcrelated 0.3, DelDup combined ped created by --includePed, 36 .jpegs rather than 72 in BAF_LRR_Plots_CNVRs.jpg for surfability, ParseCNV_GeneBased.pl option --includeIntrons added, --includeAllRedFlags option added, web version check works without error if web accessible or not, hg19 GeneRef files included in main download since typically used (including hg19_somaticRearrangement_SimFormat_AllCol.sorted which was accidentally missing in Release12), TestConcordance.pl, ConvertVCFToPennCNV.pl,ConvertQuantiSNPToPennCNV.pl scripts included, CNV_Brief.stats created as main output for quick easier to read replication with columns SNP Chr Position DelTwoTailed DupTwoTailed Cases Del Control Del Cases Dup Control Dup, NormParDel/Dup column headers renamed to DenovoDel/Dup, AnnotateCNV_MasterScanRegion.txt added for call based confidence annotation, Now Duplications are blue instead of green to aid the color blind and due to popular convention, ParseCNV_Denovo.pl added for de novo/inherited CNV Call/genotype annotation to allow for overlap parameter adjustment, father mother child CN state annotation in case PennCNV detect_cnv.pl –trio not used, and concordance between algorithms/replicates, --build bosTau7 (Cow 1-29 X) --build susScr3 (Pig 1-18 XY) now supported in addition to hg18 hg19
2013-02-05:
ParseCNVRelease_12 Posted: CHICALC.pl update to allow run from different
folder, SplitGenes_2.pl check if
conf= column, ParseCNV_QC.pl: comment.char=“” to allow # in SID, check
GCWF not all NA, if no outliers in
GCWF or CountCNV no error, No chrX summary lines used, only upon R error
is export command and R log last ten lines given, --out option enabled,
ParseCNV.pl: “package” to limit Chi2 namespace without external perl script run
for each probe (major runtime fix), check web access for content_type to
prevent error, all perl modules used are included in PerlModules folder to
limit assumptions about perl modules on user’s computer (i.e. Can't locate .pm in @INC).
2012-11-28:
ParseCNVRelease_11 Posted: --idToPath now includes two single sample plots with
CNV and flanking diploid and a sample with diploid for comparison for each CNVR
stacked into one .jpg, Sample QC ParseCNV_QC.pl module added, Gene based
approach ParseCNV_GeneBased.pl added, Exclude LRR <-6 for BafLrr plot, allow
floating point decimal for --mergePVar, semicolon delimit
TransmitUntransmitDel/Dup, --minPInclusion option added for SNPs considered in
CNVR calling, BafLrrSpecificAccess spaced for CNVR labeling, comment RedFlag
recurrent/Somatic Rearrangement, comment Exon distance, brief usage message,
allow ./ParseCNV.pl running, use Statistics::Distributions pm rather than kext
for portability and more precision (less underflow to 0), control enriched loci
reported to apply same evaluation and model case enrichment, FreqMaxPDel and
Freq>0.5 complement PenMaxP redflag to avoid exclusion based on small
subregion p=1, Sparse > 50kb inter probe distance and Freq redFlag, AvgConf
and AvgLength redFlags, ABFreqLowHigh for dups redflag, InsertPlinkPValues.pl
updated with ParseCNV updates, GIF (genomic inflation factor) for all probes
provided to gauge inflation, Simplify runtime log with no commands, SID now
needs full path and extension in .rawcnv just as provided by PennCNV and
consistency of the SID needed also in .fam and idToPath, .rawcnv needs comma in
state1,cn=0 for visualize_cnv addition.
2012-09-12:
ParseCNVRelease_10 Posted: Pdf image of multiple sample significant BAF LRR
created with --idToPath, Remove non {1-22,X,Y} chromosomes entries from UCSC
track, timeout increased to 100 for web version check to prevent content_type
undef error, hg19 GeneRef --build option, CNVRange fixed numerical sort,
cytoband annotated, ReviewedCNVRs_brief few column report generated,
scan_region and many other check output status and print error if occurs, alpha
and num map named with out to allow parallel processing by chromosome, checks
CN states prerun, --batch option for possible confounder subset of sample IDs
(see result in verbose FrequencyBatch), ChipID arrays deleted to make more
memory efficient, mergePVar and mergeDist command line options added, TDT
improved calculation and TDT drives CNVR definition (rather than case:control)
with --tdt option, complete trios are checked for available data, robust most
significant p-value in middle if p-value is strictly the same, InhDiffDel
replaced with TransmitUntransmitDel counts for TDT, check unsorted/duplicates
individual IDs or family IDs, output CNVR_DEL and CNVR_DUP (and verbose) to
temp folder to clean main output, provide active build with CNVR header.
2012-07-24:
ParseCNVRelease_9 Posted: Complicated sample IDs with “.” Supported without
control count 0 or cut off IDs in .ped, a pre-run check is done to check case
IDs in .rawcnv are all present in .fam, backtick $output=`$command` replaced
for all system($command) to ensure command completion before advance in high
memory demand runs, InsertPlinkPvalues.pl: accepts Plink p values without
.pheno as in MDS population stratification corrected logistic association
result, checks P, SNP, and one of BETA/T/OR header present.
2012-05-04:
ParseCNVRelease_8 Posted: GC threshold for red flag comment fixed, CNVRange bug
fixed, --lowHighRisk added to InsertPlinkPvalues.pl, Peninsula max P
determination fixed, UCSC quantitative trait rescaling fixed added to InsertPlinkPvalues.pl,
sorted map generated is named “Sorted_UseWithPed”, version check stored in log,
allow “.” in file name added to InsertPlinkPvalues.pl, skip -9 affected status
in .fam samples, --out output
file prefix added to InsertPlinkPvalues.pl.
2012-04-09:
ParseCNVRelease_7 Posted: Accessory function InsertPlinkPvalues.pl allows Plink
generated P values (such as .qassoc quantitative traits or any Plink association
result file) to determine CNVR boundaries, new version availability is checked
online.
2012-02-06:
ParseCNVRelease_6 Posted: Automated significant CNVR Review and Scoring
(UCSC review features), all CN state combined and each state P and OR provided
in addition to CN=0+CN=1 P and CN=3+CN=4 P, progress of CNV call loading
updated during runtime, number of probes provided in log, probe coverage UCSC
track created, --out prefix also names .map, duplicate samples warning in main
log, OR now infinity if CNV in cases and no controls, scan_region.pl now called
in PerlModules rather than embedded.
2012-01-04:
ParseCNVRelease_5 Posted: CNV .ped encoding is changed from CN=3->1
1, CN=4->1 2, other->2 2 to CN=3->1 2, CN=4->1 1, other->2 2.
This more closely resembles the CNV state frequencies defined for the deletion
.ped (CN=0->1 1, CN=1->1 2, other->2 2).
2011-09-16:
ParseCNVRelease_4 Posted:
Remove commas in numsnp and length fields. Tab delimiter in rawcnv file now
permitted. Can run from a different folder i.e. somepath/ParseCNV.pl. New
option --out output file prefix. Backtick with output stored and success
confirmation instead of system command for Sort by p-value ensure done for
large datasets. Check probes in rawcnv file are defined in map, otherwise automatically
define based on first column, and list lines in case incorrect/corrupt. On
screen prints saved to log file. Start time, end time.
2011-08-18: ParseCNVRelease_3 Posted: check for 0 before taking log. Space delimiter In Map and Fam now permitted.
2011-07-19: ParseCNVRelease_2 Posted